
Compilation of Genotype and Phenotype Data in GCDH-LOVD for Variant Classification and Further Application

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Literature Search, Submission to LOVD, and Systematic Compilation of GA-1 Genotypes and Phenotypes
2.2. Variant Evaluation
2.3. Variant Classification
2.3.1. Population and Allele Frequency Data (PM2, BA1, BS1)
2.3.2. Definition of Phenotype Specificity (PP4)
2.3.3. Functional Data
2.3.4. Use of PS4

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathogenic Criteria | ||
|---|---|---|
| Criteria | Criteria Description | Specifications |
| Very strong criteria | ||
| PVS1 | Null variant: nonsense or frameshift | Subject to SVI recommendations [34] |
| Null variant: canonical ±1,2 splice variants | Subject to SVI Splicing Subgroup recommendations [33] | |
| PM3_Very Strong | Detected in trans with a pathogenic or likely pathogenic variant
| Subject to SVI recommendations [35] Point-based system (e.g., phase confirmed: detected in three compound heterozygotes with three different variants classified as pathogenic or likely pathogenic and two homozygotes) |
| Strong criteria | ||
| PVS1_Strong | Null variant: nonsense or frameshift | Subject to SVI recommendations [34] |
| Null variant: canonical ±1,2 splice variants | Subject to SVI Splicing Subgroup recommendations [33] | |
| PM3_Strong | Detected in trans with a pathogenic or likely pathogenic variant
| Subject to SVI recommendations [35] Point-based system (e.g., phase confirmed: detected in two compound heterozygotes with two different variants classified as pathogenic or likely pathogenic) |
| PS1 | Same amino acid change as a previously established pathogenic variant regardless of nucleotide change | None |
| Same predicted splicing event as a known pathogenic or likely pathogenic variant | Subject to SVI Splicing Subgroup recommendations [33] | |
| PS3 | Well-established in vitro functional studies demonstrating a damaging effect
| Adjusted to GA-1 |
| PP3_Strong | Computational (in silico) tools predict a deleterious effect
| Subject to SVI recommendations [36] |
| Moderate criteria | ||
| PVS1_Moderate | Null variant: nonsense or frameshift | Subject to SVI recommendations [34] |
| Null variant: canonical ±1,2 splice variants | Subject to SVI Splicing Subgroup recommendations [33] | |
| Null variant: initiation codon | Subject to SVI recommendations [34] | |
| PM3 | Detected in trans with a pathogenic or likely pathogenic variant
| Subject to SVI recommendations [35] Point-based system (e.g., phase not confirmed: detected in two probands with two different variants classified as pathogenic or phase confirmed: detected in one compound heterozygote with variant classified as pathogenic or likely pathogenic) |
| PS1_Moderate | Same predicted splicing event as a pathogenic or likely pathogenic variant | Subject to SVI Splicing Subgroup recommendations [33] |
| PS4_Moderate | The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls
| Subject to ACGS 2020 guidelines [18] |
| PS3_Moderate | Well-established in vitro functional studies demonstrating a damaging effect
| Adjusted to GA-1 |
| PP3_Moderate | Computational (in silico) tools predict a deleterious effect
| Subject to SVI recommendations [36] |
| PM4 | Protein length changes as a result of in-frame deletions/insertions in a non-repeat region or stop-loss variants
| Subject to ACGS 2020 guidelines [18] |
| PM5 | Missense change at amino acid residue where a different missense change determined to be pathogenic or likely pathogenic has been seen before
| Subject to ACGS 2020 guidelines [18] |
| PP4_Moderate |
| Adjusted to GA-1 |
| Supporting criteria | ||
| PM2_Supporting | Absent from controls, or at extremely low frequency
| Adjusted to GA-1 Strength is subject to SVI recommendations [37] |
| PM3_Supporting | Detected in trans with a pathogenic or likely pathogenic variant
| Subject to SVI recommendations [35] Point-based system (e.g., phase not confirmed → detected in two probands with two different variants classified as likely pathogenic or in one proband with variant classified as pathogenic) |
| PS1_Supporting | Same predicted splicing event as a known pathogenic or likely pathogenic variant | Subject to SVI Splicing Subgroup recommendations [33] |
| PS4_Supporting | The prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls
| Subject to ACGS 2020 guidelines [18] |
| PS3_Supporting | Well-established in vitro functional studies demonstrating a damaging effect
| Adjusted to GA-1 |
| PP3_Moderate | Computational (in silico) tools predict a deleterious effect | |
| Subject to SVI recommendations [36] | |
| Subject to SVI Splicing Subgroup recommendations [33] | |
| PM4_Supporting | Protein length changes as a result of in-frame deletions/insertions in a non-repeat region or stop-loss variants
| Subject to ACGS 2020 guidelines [18] |
| PM5_Supporting | Missense change at amino acid residue where a different missense change determined to be pathogenic or likely pathogenic has been seen before
| Subject to ACGS 2020 guidelines [18] |
| PP4 |
| Adjusted to GA-1 |
| Benign criteria | ||
| Stand-alone criteria | ||
| BA1 |
| Adjusted to GA-1 |
| Strong criteria | ||
| BS1 |
| Adjusted to GA-1 |
| Supporting criteria | ||
| BP4 | Computational (in silico) tools predict no impact on gene or gene product | |
| Subject to SVI recommendations [36] | |
| Subject to SVI Splicing Subgroup recommendations [33] | |
| BP7 | Synonymous (silent) variants and intronic variants outside donor and acceptor splice regions and BP4 fulfilled | Subject to SVI Splicing Subgroup recommendations [33] |
2.4. Assessment of Variant Distribution across Different Geographic Regions
3. Results
3.1. Review of GCDH Variants
3.2. Summary of Variant Classification Using ACMG/AMP Variant Interpretation Criteria Adjusted for GCDH
3.3. Geographic Distribution of GCDH Variants
3.3.1. Global Distribution of GA-1 Patients in LOVD
3.3.2. Variant Distribution in Specific Regions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lindner, M.; Kölker, S.; Schulze, A.; Christensen, E.; Greenberg, C.R.; Hoffmann, G.F. Neonatal screening for glutaryl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2004, 27, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Boy, N.; Heringer, J.; Brackmann, R.; Bodamer, O.; Seitz, A.; Kölker, S.; Harting, I. Extrastriatal changes in patients with late-onset glutaric aciduria type I highlight the risk of long-term neurotoxicity. Orphanet J. Rare Dis. 2017, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Kölker, S.; Garbade, S.F.; Greenberg, C.R.; Leonard, J.V.; Saudubray, J.M.; Ribes, A.; Kalkanoglu, H.S.; Lund, A.M.; Merinero, B.; Wajner, M.; et al. Natural history, outcome, and treatment efficacy in children and adults with glutaryl-CoA dehydrogenase deficiency. Pediatr. Res. 2006, 59, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Gelener, P.; Severino, M.; Diker, S.; Teralı, K.; Tuncel, G.; Tuzlalı, H.; Manara, E.; Paolacci, S.; Bertelli, M.; Ergoren, M.C. Adult-onset glutaric aciduria type I: Rare presentation of a treatable disorder. Neurogenetics 2020, 21, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Boy, N.; Mengler, K.; Thimm, E.; Schiergens, K.A.; Marquardt, T.; Weinhold, N.; Marquardt, I.; Das, A.M.; Freisinger, P.; Grünert, S.C.; et al. Newborn screening: A disease-changing intervention for glutaric aciduria type 1. Ann. Neurol. 2018, 83, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Märtner, E.M.C.; Maier, E.M.; Mengler, K.; Thimm, E.; Schiergens, K.A.; Marquardt, T.; Santer, R.; Weinhold, N.; Marquardt, I.; Das, A.M.; et al. Impact of interventional and non-interventional variables on anthropometric long-term development in glutaric aciduria type 1: A national prospective multi-centre study. J. Inherit. Metab. Dis. 2021, 44, 629–638. [Google Scholar] [CrossRef]
- Boy, N.; Mengler, K.; Heringer-Seifert, J.; Hoffmann, G.F.; Garbade, S.F.; Kölker, S. Impact of newborn screening and quality of therapy on the neurological outcome in glutaric aciduria type 1: A meta-analysis. Genet. Med. 2021, 23, 13–21. [Google Scholar] [CrossRef]
- Loeber, J.G.; Platis, D.; Zetterström, R.H.; Almashanu, S.; Boemer, F.; Bonham, J.R.; Borde, P.; Brincat, I.; Cheillan, D.; Dekkers, E.; et al. Neonatal Screening in Europe Revisited: An ISNS Perspective on the Current State and Developments Since 2010. Int. J. Neonatal Screen. 2021, 7, 15. [Google Scholar] [CrossRef]
- Christensen, E.; Ribes, A.; Merinero, B.; Zschocke, J. Correlation of genotype and phenotype in glutaryl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2004, 27, 861–868. [Google Scholar] [CrossRef]
- Goodman, S.I.; Stein, D.E.; Schlesinger, S.; Christensen, E.; Schwartz, M.; Greenberg, C.R.; Elpeleg, O.N. Glutaryl-CoA dehydrogenase mutations in glutaric acidemia (type I): Review and report of thirty novel mutations. Hum. Mutat. 1998, 12, 141–144. [Google Scholar] [CrossRef]
- Yuan, Y.; Dimitrov, B.; Boy, N.; Gleich, F.; Zielonka, M.; Kölker, S. Phenotypic prediction in glutaric aciduria type 1 combining in silico and in vitro modeling with real-world data. J. Inherit. Metab. Dis. 2023, 46, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Schuurmans, I.M.E.; Dimitrov, B.; Schröter, J.; Ribes, A.; de la Fuente, R.P.; Zamora, B.; van Karnebeek, C.D.M.; Kölker, S.; Garanto, A. Exploring genotype-phenotype correlations in glutaric aciduria type 1. J. Inherit. Metab. Dis. 2023, 46, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Boy, N.; Mühlhausen, C.; Maier, E.M.; Ballhausen, D.; Baumgartner, M.R.; Beblo, S.; Burgard, P.; Chapman, K.A.; Dobbelaere, D.; Heringer-Seifert, J.; et al. Recommendations for diagnosing and managing individuals with glutaric aciduria type 1: Third revision. J. Inherit. Metab. Dis. 2023, 46, 482–519. [Google Scholar] [CrossRef] [PubMed]
- Zschocke, J.; Quak, E.; Guldberg, P.; Hoffmann, G.F. Mutation analysis in glutaric aciduria type I. J. Med. Genet. 2000, 37, 177–181. [Google Scholar] [CrossRef]
- Green, R.C.; Shah, N.; Genetti, C.A.; Yu, T.; Zettler, B.; Uveges, M.K.; Ceyhan-Birsoy, O.; Lebo, M.S.; Pereira, S.; Agrawal, P.B.; et al. Actionability of unanticipated monogenic disease risks in newborn genomic screening: Findings from the BabySeq Project. Am. J. Hum. Genet. 2023, 110, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Zastrow, D.B.; Baudet, H.; Shen, W.; Thomas, A.; Si, Y.; Weaver, M.A.; Lager, A.M.; Liu, J.; Mangels, R.; Dwight, S.S.; et al. Unique aspects of sequence variant interpretation for inborn errors of metabolism (IEM): The ClinGen IEM Working Group and the Phenylalanine Hydroxylase Gene. Hum. Mutat. 2018, 39, 1569–1580. [Google Scholar] [CrossRef]
- Ellard, S.; Baple, E.L.; Callaway, A.; Berry, I.; Forrester, N.; Turnbull, C.; Owens, M.; Eccles, D.M.; Abbs, S.; Scott, R.; et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020. Available online: https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variant-classification-v4-01-2020.pdf (accessed on 15 November 2023).
- Fokkema, I.; Kroon, M.; Lopez Hernández, J.A.; Asscheman, D.; Lugtenburg, I.; Hoogenboom, J.; den Dunnen, J.T. The LOVD3 platform: Efficient genome-wide sharing of genetic variants. Eur. J. Hum. Genet. 2021, 29, 1796–1803. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Wilcox, E.H.; Sarmady, M.; Wulf, B.; Wright, M.W.; Rehm, H.L.; Biesecker, L.G.; Abou Tayoun, A.N. Evaluating the impact of in silico predictors on clinical variant classification. Genet. Med. 2022, 24, 924–930. [Google Scholar] [CrossRef]
- Dawes, R.; Bournazos, A.M.; Bryen, S.J.; Bommireddipalli, S.; Marchant, R.G.; Joshi, H.; Cooper, S.T. SpliceVault predicts the precise nature of variant-associated mis-splicing. Nat. Genet. 2023, 55, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed]
- Tavtigian, S.V.; Greenblatt, M.S.; Harrison, S.M.; Nussbaum, R.L.; Prabhu, S.A.; Boucher, K.M.; Biesecker, L.G.; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet. Med. 2018, 20, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Tavtigian, S.V.; Harrison, S.M.; Boucher, K.M.; Biesecker, L.G. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum. Mutat. 2020, 41, 1734–1737. [Google Scholar] [CrossRef] [PubMed]
- Whiffin, N.; Minikel, E.; Walsh, R.; O’Donnell-Luria, A.H.; Karczewski, K.; Ing, A.Y.; Barton, P.J.R.; Funke, B.; Cook, S.A.; MacArthur, D.; et al. Using high-resolution variant frequencies to empower clinical genome interpretation. Genet. Med. 2017, 19, 1151–1158. [Google Scholar] [CrossRef]
- Biery, B.J.; Stein, D.E.; Morton, D.H.; Goodman, S.I. Gene structure and mutations of glutaryl-coenzyme A dehydrogenase: Impaired association of enzyme subunits that is due to an A421V substitution causes glutaric acidemia type I in the Amish. Am. J. Hum. Genet. 1996, 59, 1006–1011. [Google Scholar]
- Barroso, M.; Gertzen, M.; Puchwein-Schwepcke, A.F.; Preisler, H.; Sturm, A.; Reiss, D.D.; Danecka, M.K.; Muntau, A.C.; Gersting, S.W. Glutaryl-CoA Dehydrogenase Misfolding in Glutaric Acidemia Type 1. Int. J. Mol. Sci. 2023, 24, 13158. [Google Scholar] [CrossRef]
- Schmiesing, J.; Lohmöller, B.; Schweizer, M.; Tidow, H.; Gersting, S.W.; Muntau, A.C.; Braulke, T.; Mühlhausen, C. Disease-causing mutations affecting surface residues of mitochondrial glutaryl-CoA dehydrogenase impair stability, heteromeric complex formation and mitochondria architecture. Hum. Mol. Genet. 2017, 26, 538–551. [Google Scholar] [CrossRef]
- Westover, J.B.; Goodman, S.I.; Frerman, F.E. Pathogenic mutations in the carboxyl-terminal domain of glutaryl-CoA dehydrogenase: Effects on catalytic activity and the stability of the tetramer. Mol. Genet. Metab. 2003, 79, 245–256. [Google Scholar] [CrossRef]
- Keyser, B.; Mühlhausen, C.; Dickmanns, A.; Christensen, E.; Muschol, N.; Ullrich, K.; Braulke, T. Disease-causing missense mutations affect enzymatic activity, stability and oligomerization of glutaryl-CoA dehydrogenase (GCDH). Hum. Mol. Genet. 2008, 17, 3854–3863. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, M.; Paschke, R.; Rao, K.S.; Frerman, F.E.; Kim, J.J. Crystal structures of human glutaryl-CoA dehydrogenase with and without an alternate substrate: Structural bases of dehydrogenation and decarboxylation reactions. Biochemistry 2004, 43, 9674–9684. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; de la Hoya, M.; Wiggins, G.A.R.; Lindy, A.; Vincent, L.M.; Parsons, M.T.; Canson, D.M.; Bis-Brewer, D.; Cass, A.; Tchourbanov, A.; et al. Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: Recommendations from the ClinGen SVI Splicing Subgroup. Am. J. Hum. Genet. 2023, 110, 1046–1067. [Google Scholar] [CrossRef]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M.; ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef]
- Sequence Variant Interpretation Working Group. SVI Recommendation for in Trans Criterion (PM3)—Version 1.0. Available online: https://clinicalgenome.org/site/assets/files/3717/svi_proposal_for_pm3_criterion_-_version_1.pdf (accessed on 19 November 2023).
- Pejaver, V.; Byrne, A.B.; Feng, B.J.; Pagel, K.A.; Mooney, S.D.; Karchin, R.; O’Donnell-Luria, A.; Harrison, S.M.; Tavtigian, S.V.; Greenblatt, M.S.; et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am. J. Hum. Genet. 2022, 109, 2163–2177. [Google Scholar] [CrossRef]
- Sequence Variant Interpretation Working Group. ClinGen Sequence Variant Interpretation Recommendation for PM2—Version 1.0. Available online: https://clinicalgenome.org/site/assets/files/5182/pm2_-_svi_recommendation_-_approved_sept2020.pdf (accessed on 15 November 2023).
- Liang, L.; Zhang, H.; Qiu, W.; Ye, J.; Xu, F.; Gong, Z.; Gu, X.; Han, L. Evaluation of the Clinical, Biochemical, Neurological, and Genetic Presentations of Glutaric Aciduria Type 1 in Patients From China. Front. Genet. 2021, 12, 702374. [Google Scholar] [CrossRef]
- Georgiou, T.; Nicolaidou, P.; Hadjichristou, A.; Ioannou, R.; Dionysiou, M.; Siama, E.; Chappa, G.; Anastasiadou, V.; Drousiotou, A. Molecular analysis of Cypriot patients with Glutaric aciduria type I: Identification of two novel mutations. Clin. Biochem. 2014, 47, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Tamhankar, P.M.; Vasudevan, L.; Kondurkar, P.; Niazi, S.; Christopher, R.; Solanki, D.; Dholakia, P.; Muranjan, M.; Kamate, M.; Kalane, U.; et al. Clinical Characteristics, Molecular Profile, and Outcomes in Indian Patients with Glutaric Aciduria Type 1. J. Pediatr. Genet. 2021, 10, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Zafeiriou, D.I.; Zschocke, J.; Augoustidou-Savvopoulou, P.; Mauromatis, I.; Sewell, A.; Kontopoulos, E.; Katzos, G.; Hoffmann, G.F. Atypical and variable clinical presentation of glutaric aciduria type I. Neuropediatrics 2000, 31, 303–306. [Google Scholar] [CrossRef]
- Harting, I.; Neumaier-Probst, E.; Seitz, A.; Maier, E.M.; Assmann, B.; Baric, I.; Troncoso, M.; Mühlhausen, C.; Zschocke, J.; Boy, N.P.; et al. Dynamic changes of striatal and extrastriatal abnormalities in glutaric aciduria type I. Brain 2009, 132, 1764–1782. [Google Scholar] [CrossRef]
- Wang, Q.; Li, X.; Ding, Y.; Liu, Y.; Song, J.; Yang, Y. Clinical and mutational spectra of 23 Chinese patients with glutaric aciduria type 1. Brain Dev. 2014, 36, 813–822. [Google Scholar] [CrossRef]
- Bross, P.; Frederiksen, J.B.; Bie, A.S.; Hansen, J.; Palmfeldt, J.; Nielsen, M.N.; Duno, M.; Lund, A.M.; Christensen, E. Heterozygosity for an in-frame deletion causes glutaryl-CoA dehydrogenase deficiency in a patient detected by newborn screening: Investigation of the effect of the mutant allele. J. Inherit. Metab. Dis. 2012, 35, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Radha Rama Devi, A.; Ramesh, V.A.; Nagarajaram, H.A.; Satish, S.P.; Jayanthi, U.; Lingappa, L. Spectrum of mutations in Glutaryl-CoA dehydrogenase gene in glutaric aciduria type I—Study from South India. Brain Dev. 2016, 38, 54–60. [Google Scholar] [CrossRef] [PubMed]
- David, A.; Sternberg, M.J.E. Protein structure-based evaluation of missense variants: Resources, challenges and future directions. Curr. Opin. Struct. Biol. 2023, 80, 102600. [Google Scholar] [CrossRef] [PubMed]
- Scheller, R.; Stein, A.; Nielsen, S.V.; Marin, F.I.; Gerdes, A.M.; Di Marco, M.; Papaleo, E.; Lindorff-Larsen, K.; Hartmann-Petersen, R. Toward mechanistic models for genotype-phenotype correlations in phenylketonuria using protein stability calculations. Hum. Mutat. 2019, 40, 444–457. [Google Scholar] [CrossRef]
- Caswell, R.C.; Owens, M.M.; Gunning, A.C.; Ellard, S.; Wright, C.F. Using Structural Analysis In Silico to Assess the Impact of Missense Variants in MEN1. J. Endocr. Soc. 2019, 3, 2258–2275. [Google Scholar] [CrossRef]
- Therrell, B.L., Jr.; Lloyd-Puryear, M.A.; Ohene-Frempong, K.; Ware, R.E.; Padilla, C.D.; Ambrose, E.E.; Barkat, A.; Ghazal, H.; Kiyaga, C.; Mvalo, T.; et al. Empowering newborn screening programs in African countries through establishment of an international collaborative effort. J. Community Genet. 2020, 11, 253–268. [Google Scholar] [CrossRef]
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.; Adams, J. Current status of newborn screening worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef]
- Jongeneel, C.V.; Kotze, M.J.; Bhaw-Luximon, A.; Fadlelmola, F.M.; Fakim, Y.J.; Hamdi, Y.; Kassim, S.K.; Kumuthini, J.; Nembaware, V.; Radouani, F.; et al. A View on Genomic Medicine Activities in Africa: Implications for Policy. Front. Genet. 2022, 13, 769919. [Google Scholar] [CrossRef]
- Poletto, E.; Pasqualim, G.; Giugliani, R.; Matte, U.; Baldo, G. Worldwide distribution of common IDUA pathogenic variants. Clin. Genet. 2018, 94, 95–102. [Google Scholar] [CrossRef]
- Pereira, L.; Mutesa, L.; Tindana, P.; Ramsay, M. African genetic diversity and adaptation inform a precision medicine agenda. Nat. Rev. Genet. 2021, 22, 284–306. [Google Scholar] [CrossRef]
- Bertranpetit, J.; Calafell, F. Genetic and geographical variability in cystic fibrosis: Evolutionary considerations. Ciba Found. Symp. 1996, 197, 97–114, discussion 114–118. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, C.R.; Prasad, A.N.; Dilling, L.A.; Thompson, J.R.; Haworth, J.C.; Martin, B.; Wood-Steiman, P.; Seargeant, L.E.; Seifert, B.; Booth, F.A.; et al. Outcome of the first 3-years of a DNA-based neonatal screening program for glutaric acidemia type 1 in Manitoba and northwestern Ontario, Canada. Mol. Genet. Metab. 2002, 75, 70–78. [Google Scholar] [CrossRef]
- Greenberg, C.R.; Reimer, D.; Singal, R.; Triggs-Raine, B.; Chudley, A.E.; Dilling, L.A.; Philipps, S.; Haworth, J.C.; Seargeant, L.E.; Goodman, S.I. A G-to-T transversion at the +5 position of intron 1 in the glutaryl CoA dehydrogenase gene is associated with the Island Lake variant of glutaric acidemia type I. Hum. Mol. Genet. 1995, 4, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Haworth, J.C.; Booth, F.A.; Chudley, A.E.; deGroot, G.W.; Dilling, L.A.; Goodman, S.I.; Greenberg, C.R.; Mallory, C.J.; McClarty, B.M.; Seshia, S.S.; et al. Phenotypic variability in glutaric aciduria type I: Report of fourteen cases in five Canadian Indian kindreds. J. Pediatr. 1991, 118, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.B.; Prasad, A.N.; Frosk, P.; Sauer, S.; Kölker, S.; Greenberg, C.R.; Del Bigio, M.R. Neuropathological, biochemical and molecular findings in a glutaric acidemia type 1 cohort. Brain 2005, 128, 711–722. [Google Scholar] [CrossRef]
- Haworth, J.C.; Dilling, L.A.; Seargeant, L.E. Increased prevalence of hereditary metabolic diseases among native Indians in Manitoba and northwestern Ontario. Can. Med. Assoc. J. 1991, 145, 123–129. [Google Scholar]

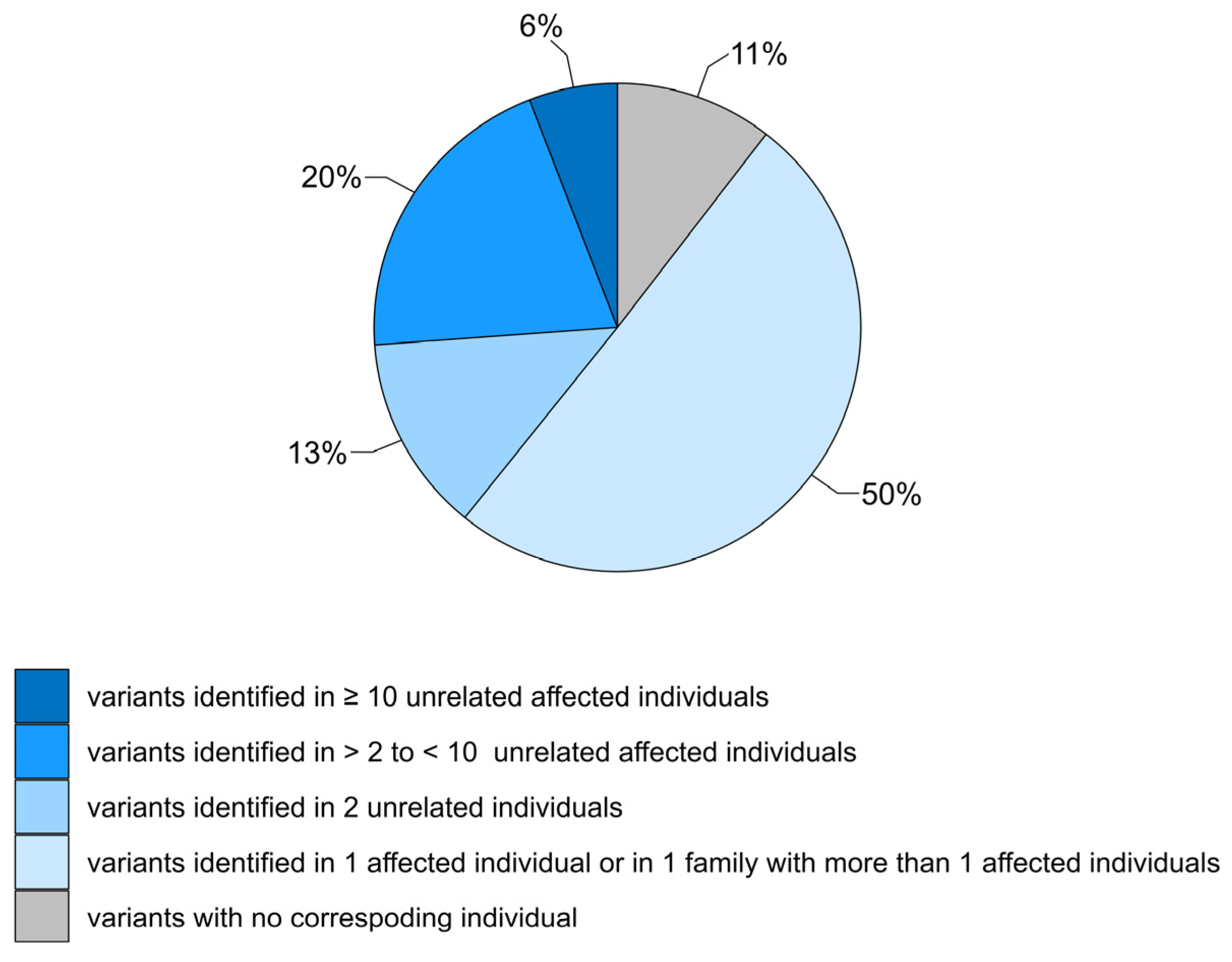


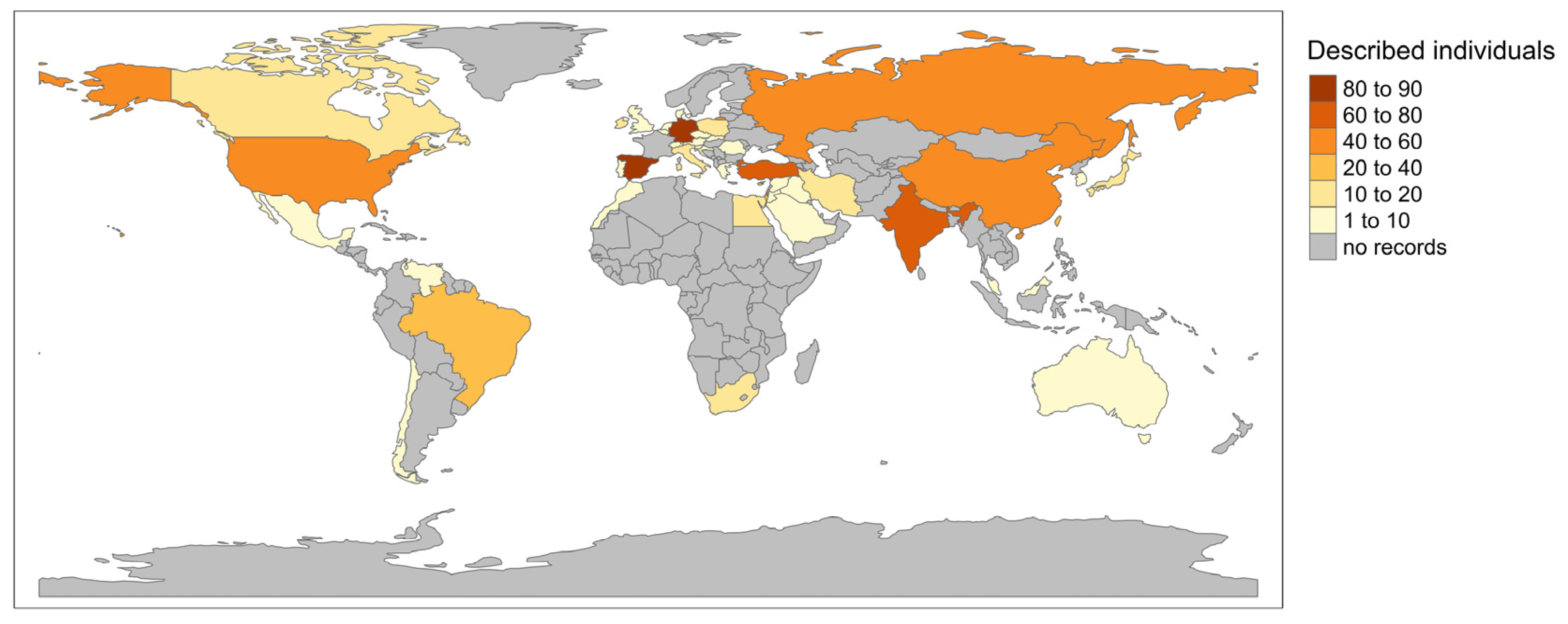
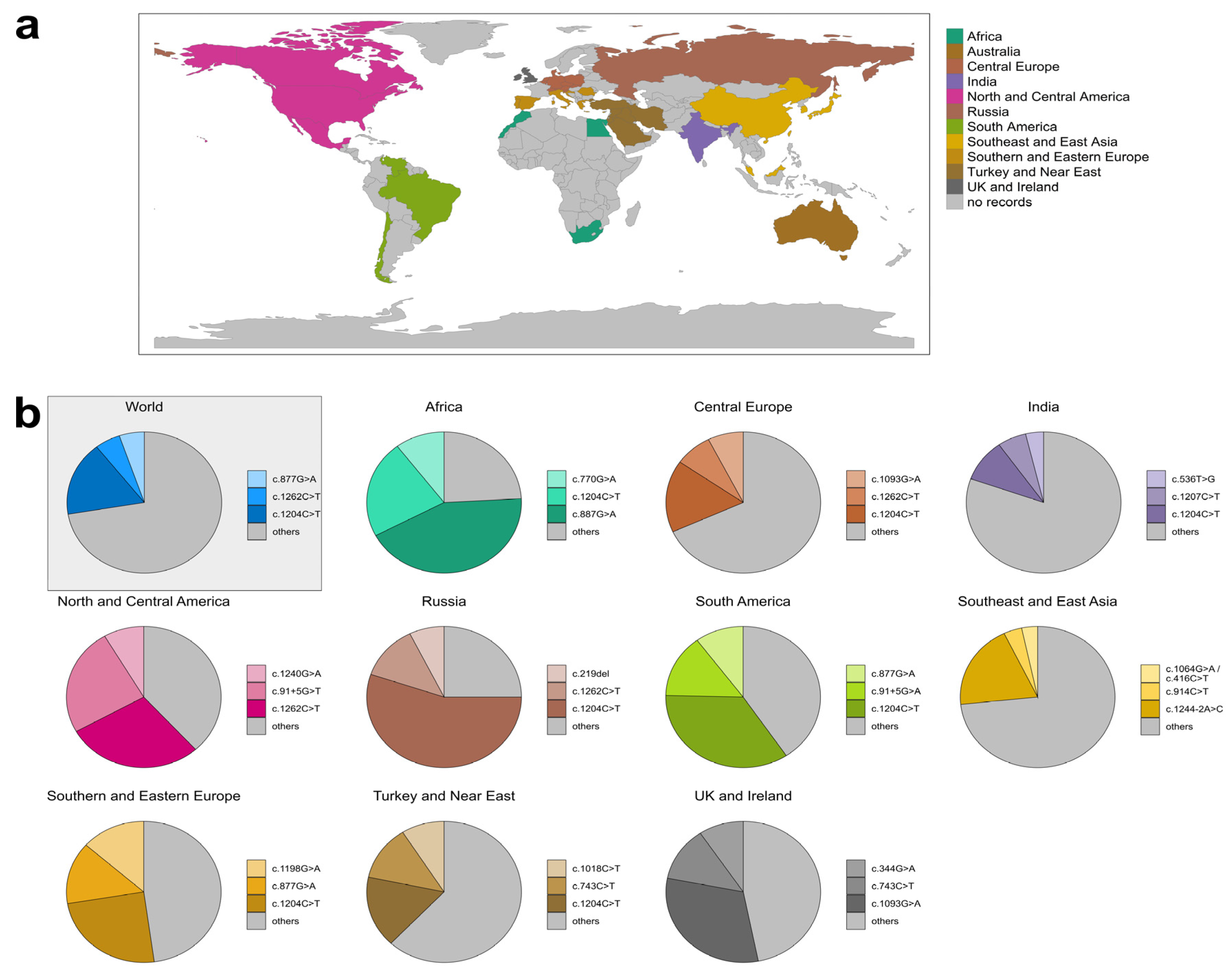
| Classification | Variants with Patient Data | Variants without Patient Data |
|---|---|---|
| Pathogenic | 93 | 0 |
| Likely pathogenic | 105 | 7 |
| VUS (hot) | 22 | 3 |
| VUS (warm) | 21 | 1 |
| VUS (tepid) | 20 | 5 |
| VUS (cool) | 8 | 1 |
| VUS (cold) | 4 | 1 |
| VUS (ice-cold) | 0 | 0 |
| Likely benign | 0 | 6 |
| Benign | 1 | 8 |
| Total | 274 | 32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tibelius, A.; Evers, C.; Oeser, S.; Rinke, I.; Jauch, A.; Hinderhofer, K. Compilation of Genotype and Phenotype Data in GCDH-LOVD for Variant Classification and Further Application. Genes 2023, 14, 2218. https://doi.org/10.3390/genes14122218
Tibelius A, Evers C, Oeser S, Rinke I, Jauch A, Hinderhofer K. Compilation of Genotype and Phenotype Data in GCDH-LOVD for Variant Classification and Further Application. Genes. 2023; 14(12):2218. https://doi.org/10.3390/genes14122218
Chicago/Turabian StyleTibelius, Alexandra, Christina Evers, Sabrina Oeser, Isabelle Rinke, Anna Jauch, and Katrin Hinderhofer. 2023. "Compilation of Genotype and Phenotype Data in GCDH-LOVD for Variant Classification and Further Application" Genes 14, no. 12: 2218. https://doi.org/10.3390/genes14122218