

Influence of Model Structures on Predictors of Protein Stability Changes from Single-Point Mutations

 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Predictors
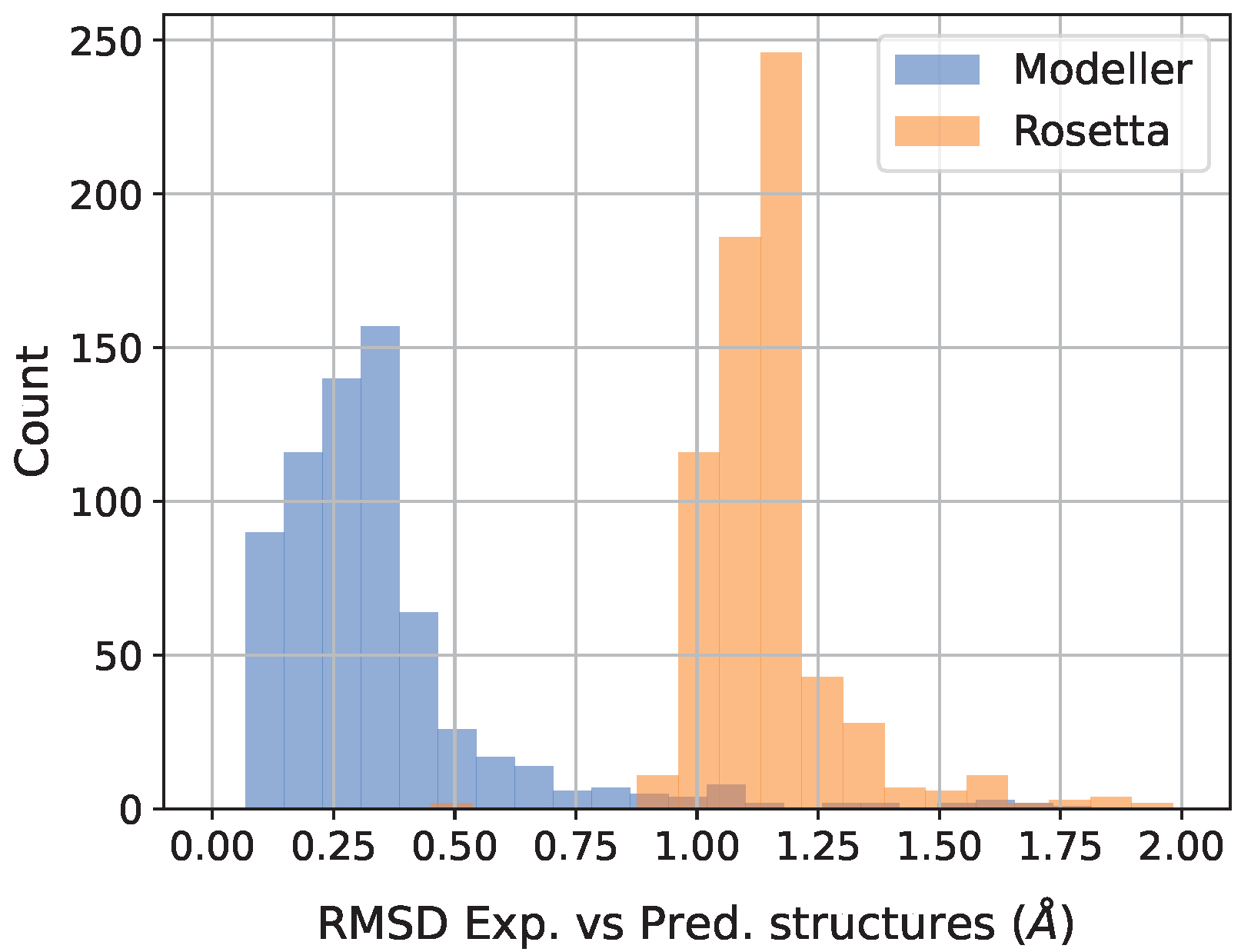
2.2. Protein 3D-Structure Models
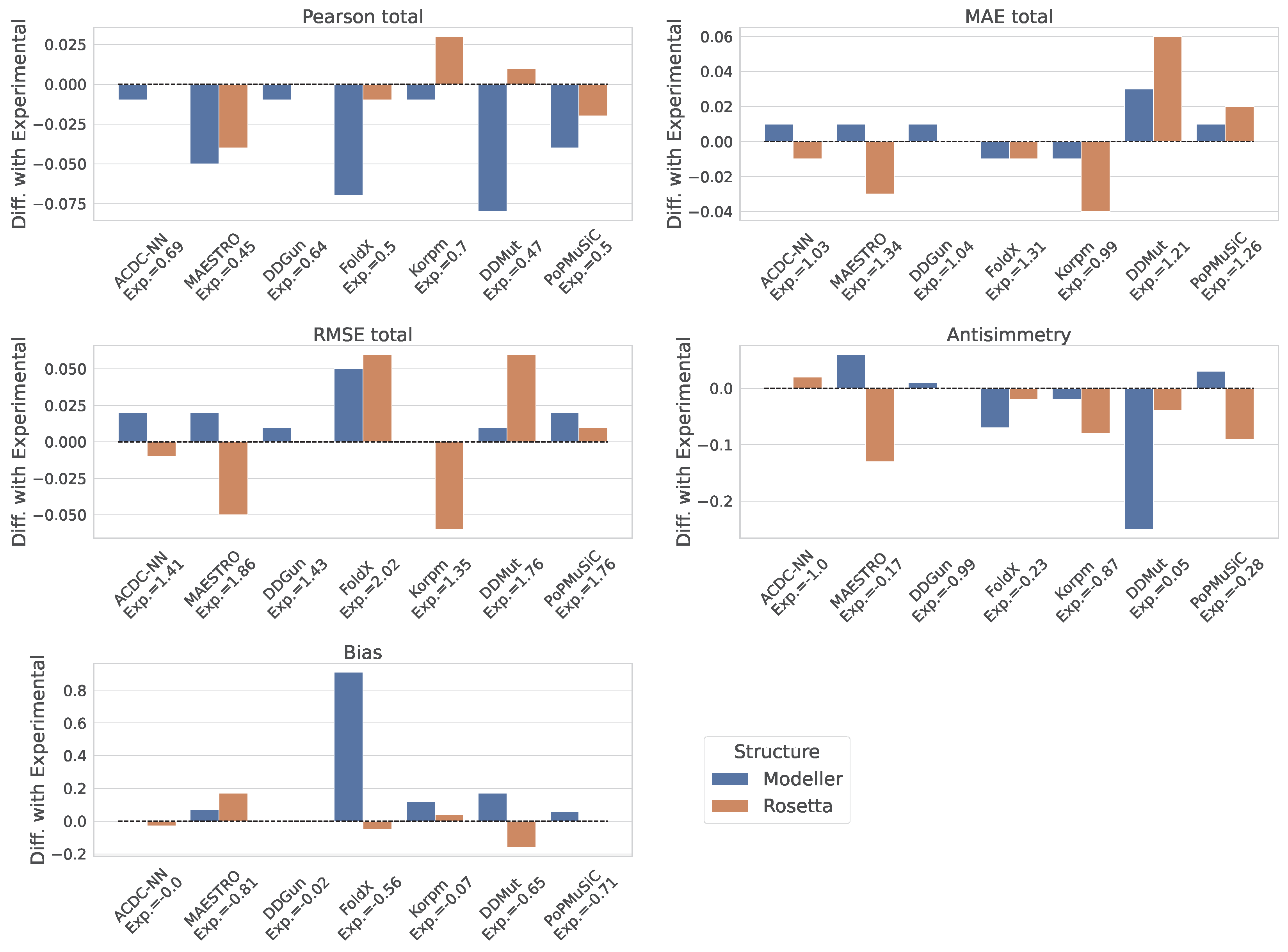
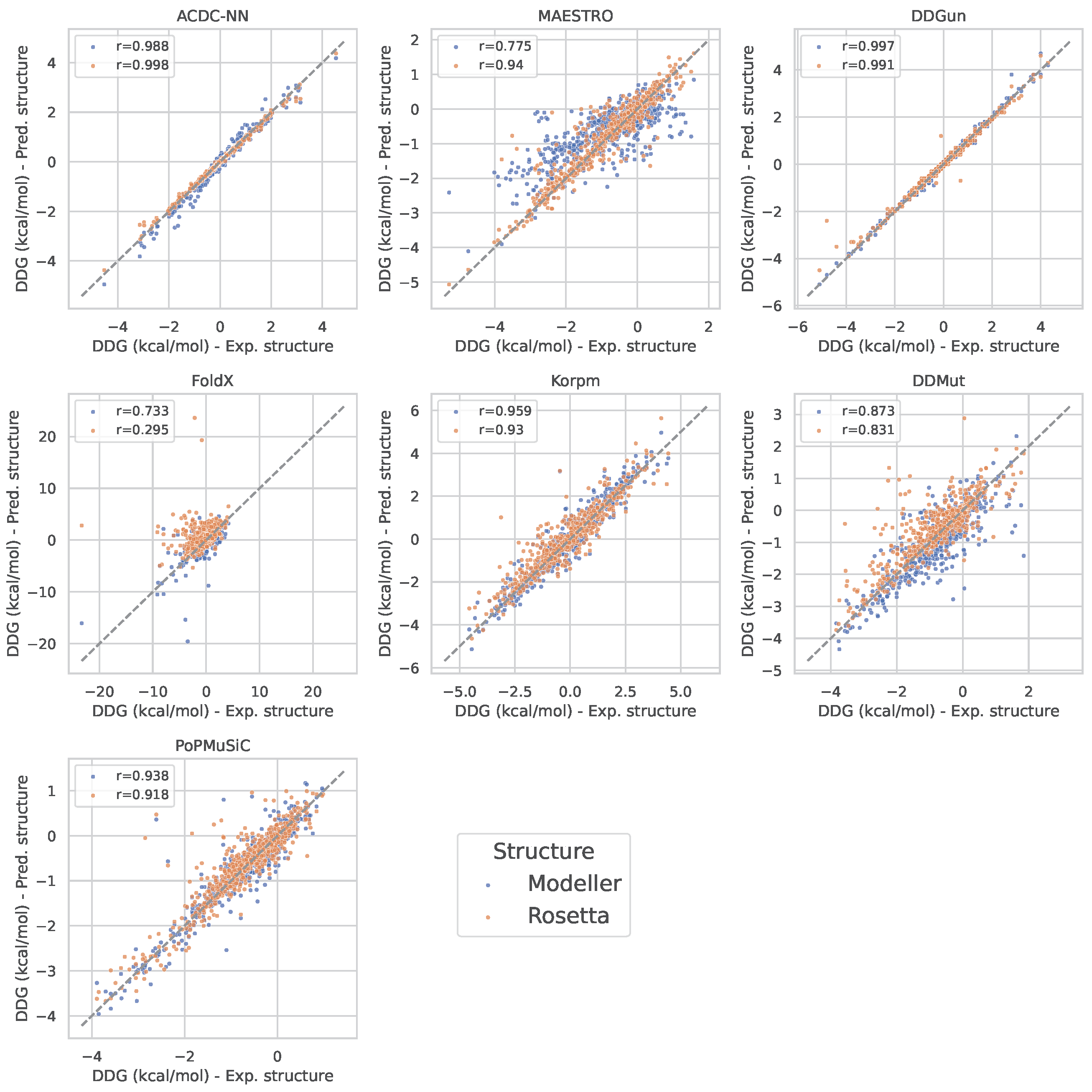
2.3. Assessment of Predictors’ Performance
3. Results
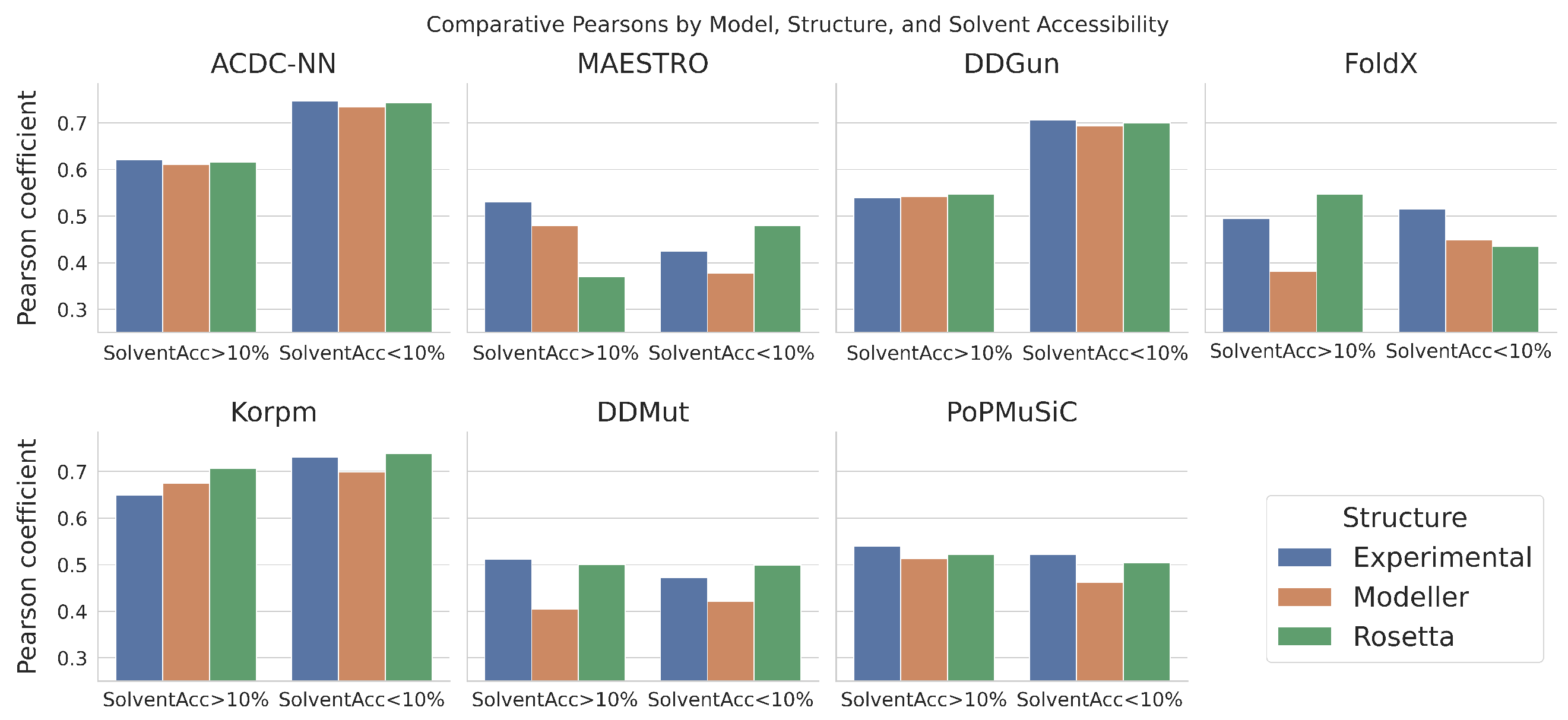
Comparative Analysis Based on Solvent Accessibility
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Casadio, R.; Vassura, M.; Tiwari, S.; Fariselli, P.; Luigi Martelli, P. Correlating disease-related mutations to their effect on protein stability: A large-scale analysis of the human proteome. Hum. Mutat. 2011, 32, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Alexov, E. Most monogenic disorders are caused by mutations altering protein folding free energy. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Nakano, S.i.; Sugimoto, N. The structural stability and catalytic activity of DNA and RNA oligonucleotides in the presence of organic solvents. Biophys. Rev. 2016, 8, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, J.L.; Schmidt, F.H. The role of mRNA and protein stability in gene expression. FASEB J. 1989, 3, 2360–2370. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.M.; Lu, Y.E.; Vendruscolo, M.; Lio’, P.; Blundell, T.L. Prediction by graph theoretic measures of structural effects in proteins arising from non-synonymous single nucleotide polymorphisms. PLoS Comput. Biol. 2008, 4, e1000135. [Google Scholar] [CrossRef] [PubMed]
- Compiani, M.; Capriotti, E. Computational and theoretical methods for protein folding. Biochemistry 2013, 52, 8601–8624. [Google Scholar] [CrossRef] [PubMed]
- Martelli, P.L.; Fariselli, P.; Savojardo, C.; Babbi, G.; Aggazio, F.; Casadio, R. Large scale analysis of protein stability in OMIM disease related human protein variants. BMC Genom. 2016, 17, 239–247. [Google Scholar] [CrossRef]
- Hartl, F.U. Protein misfolding diseases. Annu. Rev. Biochem. 2017, 86, 21–26. [Google Scholar] [CrossRef]
- Lam, S.D.; Das, S.; Sillitoe, I.; Orengo, C. An overview of comparative modelling and resources dedicated to large-scale modelling of genome sequences. Acta Crystallogr. D Struct. Biol. 2017, 73 Pt 8, 628–640. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Hameduh, T.; Haddad, Y.; Adam, V.; Heger, Z. Homology modeling in the time of collective and artificial intelligence. Comput. Struct. Biotechnol. J. 2020, 18, 3494–3506. [Google Scholar] [CrossRef] [PubMed]
- Marabotti, A.; Scafuri, B.; Facchiano, A. Predicting the stability of mutant proteins by computational approaches: An overview. Brief. Bioinform. 2021, 22, bbaa074. [Google Scholar] [CrossRef] [PubMed]
- Pancotti, C.; Benevenuta, S.; Birolo, G.; Alberini, V.; Repetto, V.; Sanavia, T.; Capriotti, E.; Fariselli, P. Predicting protein stability changes upon single-point mutation: A thorough comparison of the available tools on a new dataset. Brief. Bioinform. 2022, 23, bbab555. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Ascher, D.B.; Blundell, T.L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Buß, O.; Rudat, J.; Ochsenreither, K. FoldX as protein engineering tool: Better than random based approaches? Comput. Struct. Biotechnol. J. 2018, 16, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Laimer, J.; Hofer, H.; Fritz, M.; Wegenkittl, S.; Lackner, P. MAESTRO-multi agent stability prediction upon point mutations. BMC Bioinform. 2015, 16, 116. [Google Scholar] [CrossRef]
- Dehouck, Y.; Kwasigroch, J.M.; Gilis, D.; Rooman, M. PoPMuSiC 2.1: A web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinform. 2011, 12, 151. [Google Scholar] [CrossRef]
- Hernández, I.M.; Dehouck, Y.; Bastolla, U.; López-Blanco, J.R.; Chacón, P. Predicting protein stability changes upon mutation using a simple orientational potential. Bioinformatics 2023, 39, btad011. [Google Scholar] [CrossRef]
- Hou, Q.; Pucci, F.; Ancien, F.; Kwasigroch, J.M.; Bourgeas, R.; Rooman, M. SWOTein: A structure-based approach to predict stability strengths and weaknesses of prOTEINs. Bioinformatics 2021, 37, 1963–1971. [Google Scholar] [CrossRef]
- Savojardo, C.; Fariselli, P.; Martelli, P.L.; Casadio, R. INPS-MD: A web server to predict stability of protein variants from sequence and structure. Bioinformatics 2016, 32, 2542–2544. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Pan, Q.; Pires, D.E.; Rodrigues, C.H.; Ascher, D.B. DDMut: Predicting effects of mutations on protein stability using deep learning. Nucleic Acids Res. 2023, 51, W122–W128. [Google Scholar] [CrossRef] [PubMed]
- Pucci, F.; Schwersensky, M.; Rooman, M. Artificial intelligence challenges for predicting the impact of mutations on protein stability. Curr. Opin. Struct. Biol. 2022, 72, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Ancien, F.; Pucci, F.; Godfroid, M.; Rooman, M. Prediction and interpretation of deleterious coding variants in terms of protein structural stability. Sci. Rep. 2018, 8, 4480. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, Y.T.; Capra, J.A.; Gerstein, M.B. Predicting changes in protein thermodynamic stability upon point mutation with deep 3D convolutional neural networks. PLoS Comput. Biol. 2020, 16, e1008291. [Google Scholar]
- Yang, Y.; Chen, B.; Tan, G.; Vihinen, M.; Shen, B. Structure-based prediction of the effects of a missense variant on protein stability. Amino Acids 2013, 44, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ding, X.; Zhu, G.; Niroula, A.; Lv, Q.; Vihinen, M. ProTstab–predictor for cellular protein stability. BMC Genom. 2019, 20, 804. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, J.; Zeng, L.; Vihinen, M. ProTstab2 for Prediction of Protein Thermal Stabilities. Int. J. Mol. Sci. 2022, 23, 10798. [Google Scholar] [CrossRef]
- Benevenuta, S.; Pancotti, C.; Fariselli, P.; Birolo, G.; Sanavia, T. An antisymmetric neural network to predict free energy changes in protein variants. J. Phys. D Appl. Phys. 2021, 54, 245403. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. In Functional Genomics: Methods and Protocols; Humana Press: New York, NY, USA, 2017; pp. 39–54. [Google Scholar]
- Kellogg, E.H.; Leaver-Fay, A.; Baker, D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins Struct. Funct. Bioinform. 2011, 79, 830–838. [Google Scholar] [CrossRef]
- Pan, Q.; Nguyen, T.B.; Ascher, D.B.; Pires, D.E.V. Systematic evaluation of computational tools to predict the effects of mutations on protein stability in the absence of experimental structures. Brief. Bioinform. 2022, 23, bbac025. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Minton, K. Predicting variant pathogenicity with AlphaMissense. Nat. Rev. Genet. 2023, 24, 804. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulytė, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [CrossRef] [PubMed]
- York, D.M. Modern Alchemical Free Energy Methods for Drug Discovery Explained. ACS Phys. Chem. Au 2023, 3, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Pak, M.A.; Markhieva, K.A.; Novikova, M.S.; Petrov, D.S.; Vorobyev, I.S.; Maksimova, E.S.; Kondrashov, F.A.; Ivankov, D.N. Using AlphaFold to predict the impact of single mutations on protein stability and function. PLoS ONE 2023, 18, e0282689. [Google Scholar] [CrossRef]
- Pucci, F.; Bernaerts, K.V.; Kwasigroch, J.M.; Rooman, M. Quantification of biases in predictions of protein stability changes upon mutations. Bioinformatics 2018, 34, 3659–3665. [Google Scholar] [CrossRef]
- Montanucci, L.; Capriotti, E.; Birolo, G.; Benevenuta, S.; Pancotti, C.; Lal, D.; Fariselli, P. DDGun: An untrained predictor of protein stability changes upon amino acid variants. Nucleic Acids Res. 2022, 50, W222–W227. [Google Scholar] [CrossRef]
- Song, Y.; DiMaio, F.; Wang, R.Y.R.; Kim, D.; Miles, C.; Brunette, T.J.; Thompson, J.; Baker, D. High-resolution comparative modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACDC-NN | 0.69 | 0.61 | 0.61 | 1.03 | 1.03 | 1.03 | 1.41 | 1.41 | 1.41 | −1.00 | 0.00 |
| MAESTRO | 0.45 | 0.59 | 0.22 | 1.34 | 0.90 | 1.78 | 1.86 | 1.27 | 2.30 | −0.17 | −0.81 |
| DDGun | 0.64 | 0.55 | 0.53 | 1.04 | 1.02 | 1.05 | 1.43 | 1.41 | 1.45 | −0.99 | −0.02 |
| FoldX | 0.50 | 0.57 | 0.38 | 1.31 | 1.16 | 1.47 | 2.02 | 1.89 | 2.14 | −0.23 | −0.56 |
| Korpm | 0.70 | 0.57 | 0.49 | 0.99 | 0.96 | 1.02 | 1.35 | 1.30 | 1.39 | −0.87 | −0.07 |
| DDMut | 0.47 | 0.81 | −0.02 | 1.21 | 0.67 | 1.76 | 1.76 | 0.94 | 2.30 | 0.05 | −0.65 |
| PoPMuSiC | 0.50 | 0.63 | 0.25 | 1.26 | 0.86 | 1.66 | 1.76 | 1.21 | 2.18 | −0.28 | −0.71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rollo, C.; Pancotti, C.; Birolo, G.; Rossi, I.; Sanavia, T.; Fariselli, P. Influence of Model Structures on Predictors of Protein Stability Changes from Single-Point Mutations. Genes 2023, 14, 2228. https://doi.org/10.3390/genes14122228
Rollo C, Pancotti C, Birolo G, Rossi I, Sanavia T, Fariselli P. Influence of Model Structures on Predictors of Protein Stability Changes from Single-Point Mutations. Genes. 2023; 14(12):2228. https://doi.org/10.3390/genes14122228
Chicago/Turabian StyleRollo, Cesare, Corrado Pancotti, Giovanni Birolo, Ivan Rossi, Tiziana Sanavia, and Piero Fariselli. 2023. "Influence of Model Structures on Predictors of Protein Stability Changes from Single-Point Mutations" Genes 14, no. 12: 2228. https://doi.org/10.3390/genes14122228
APA StyleRollo, C., Pancotti, C., Birolo, G., Rossi, I., Sanavia, T., & Fariselli, P. (2023). Influence of Model Structures on Predictors of Protein Stability Changes from Single-Point Mutations. Genes, 14(12), 2228. https://doi.org/10.3390/genes14122228