
Integrated Metabolomic and Transcriptomic Analysis of Puerarin Biosynthesis in Pueraria montana var. thomsonii at Different Growth Stages


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Processing of Samples and Procedures of HPLC
2.3. RNA Extraction and RNA Sequencing
2.4. UPLC–MS/MS-Based Widely Targeted Metabolomic Analysis
2.5. Analysis of Differential Accumulation Metabolites and Differentially Expressed Genes
2.6. Kyoto Encyclopedia of Genes and Genomes Annotation and Enrichment Analysis
2.7. Identification of Structural Genes in the Puerarin Biosynthesis Pathway
2.8. Phylogenetic Analysis of UGTs in the Puerarin Biosynthesis Pathway
2.9. Quantitative Real-Time PCR
3. Results
3.1. Determination of Puerarin in P. montana var. thomsonii at Different Growth Stages via HPLC
3.2. Metabolomic Profiling and Differentially Accumulated Metabolite Enrichment of P. montana var. thomsonii at Different Growth Stages
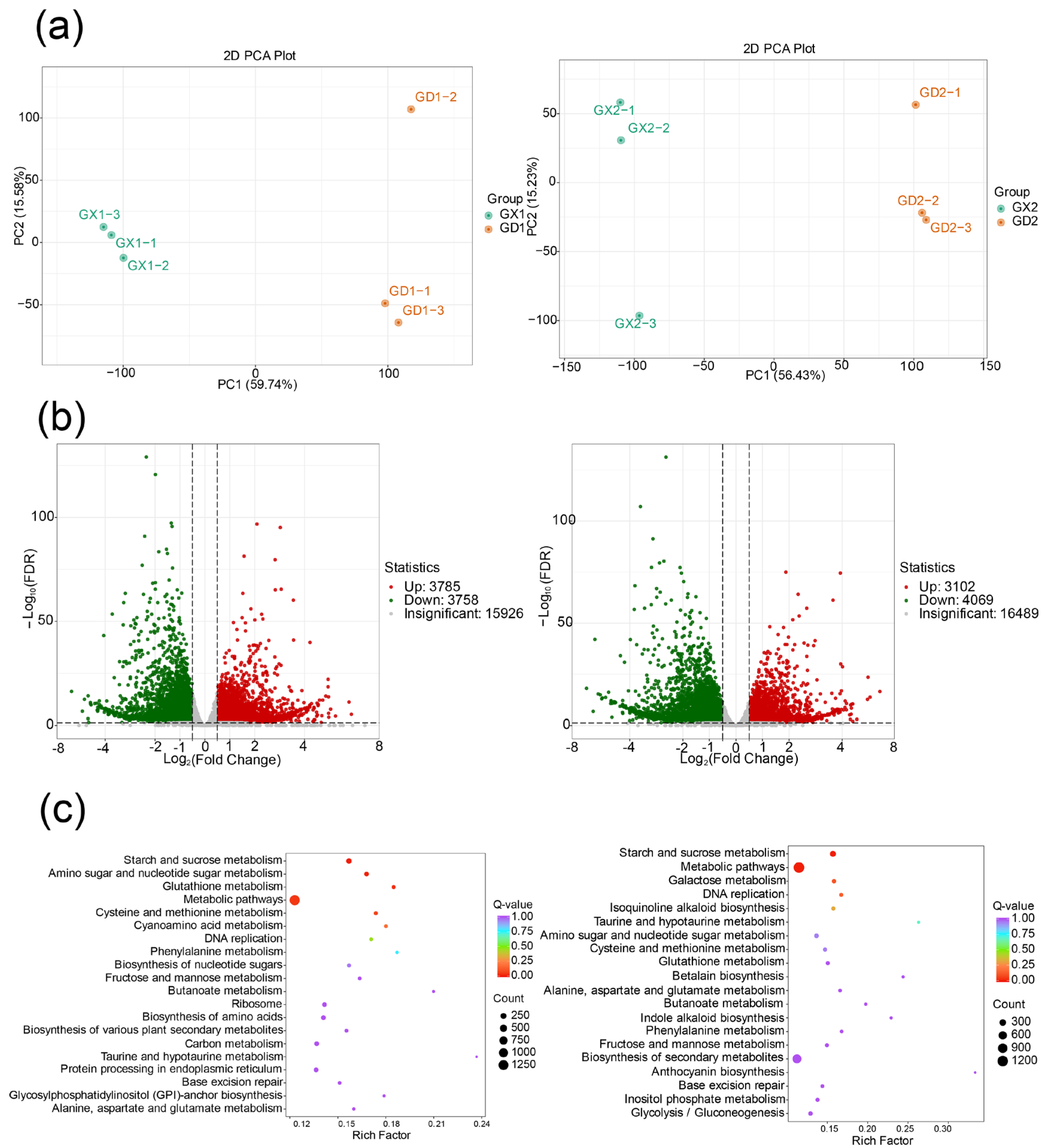
3.3. Transcriptomic Profiling and Differentially Expressed Gene Screening of P. montana var. thomsonii at Different Growth Stages
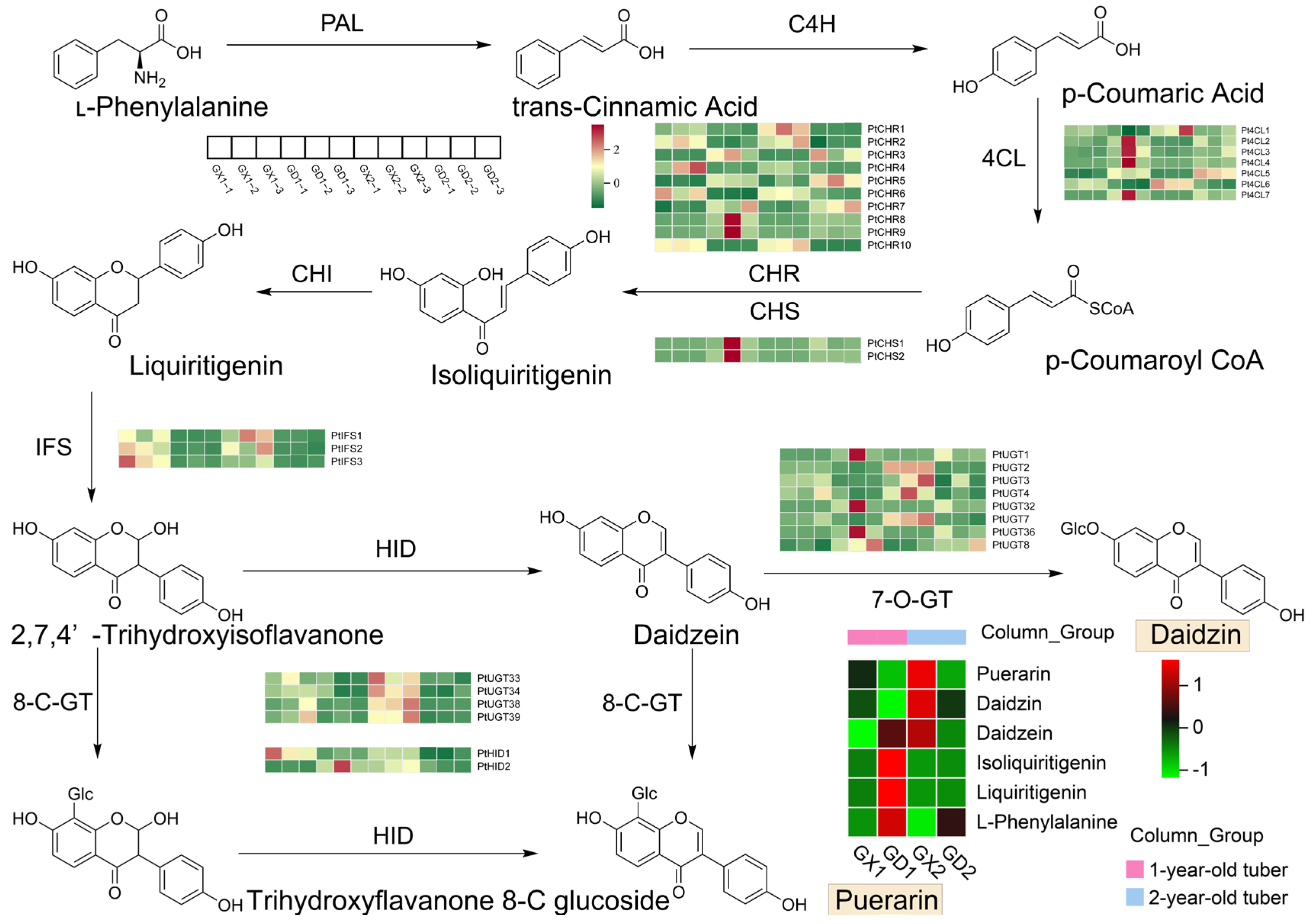
3.4. Analysis of Metabolites and Differentially Expressed Genes Involved in the Puerarin Biosynthesis Pathway
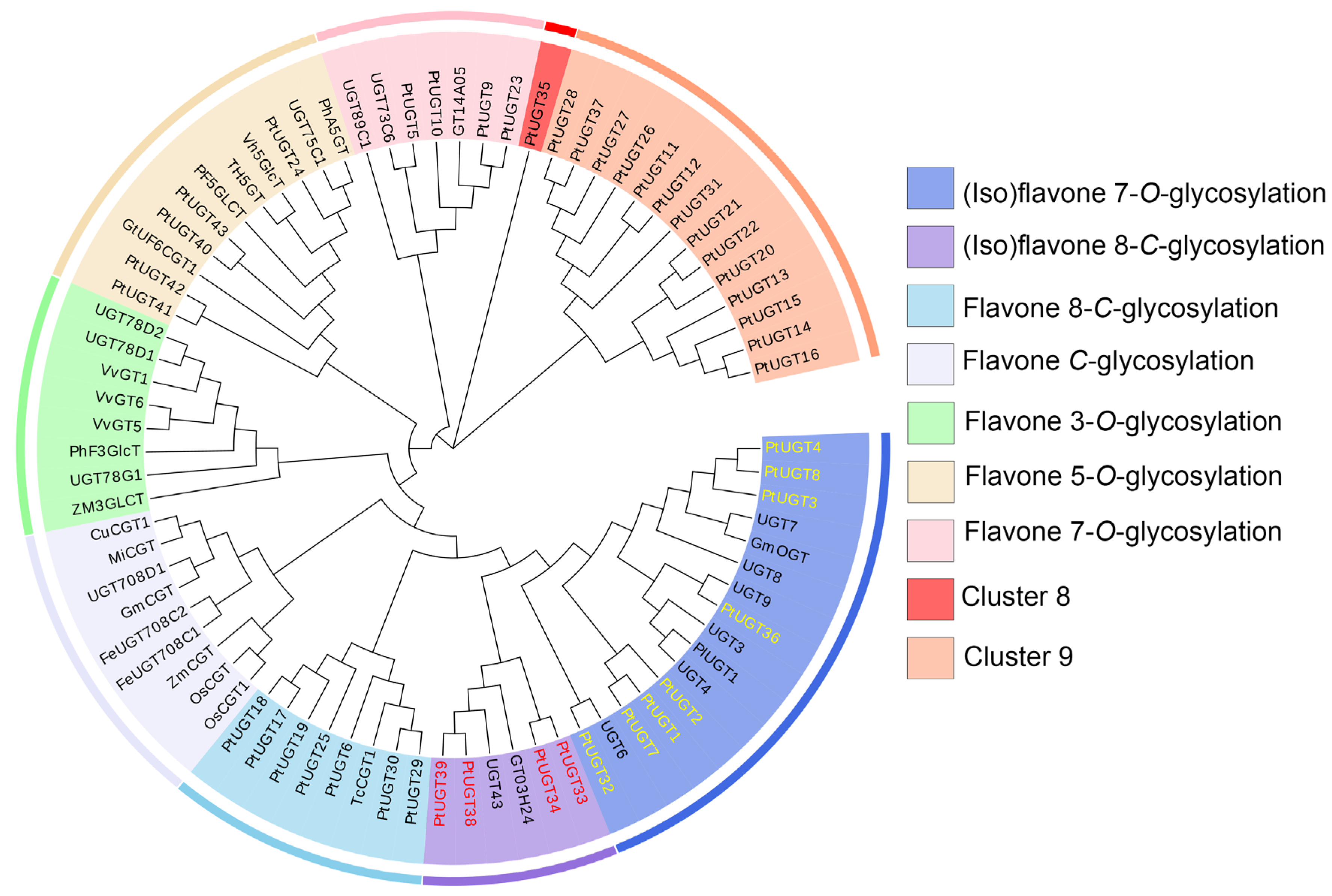
3.5. Phylogenetic Analysis of Candidate UGTs
3.6. Co-Expression Analysis of Important Genes and Metabolites
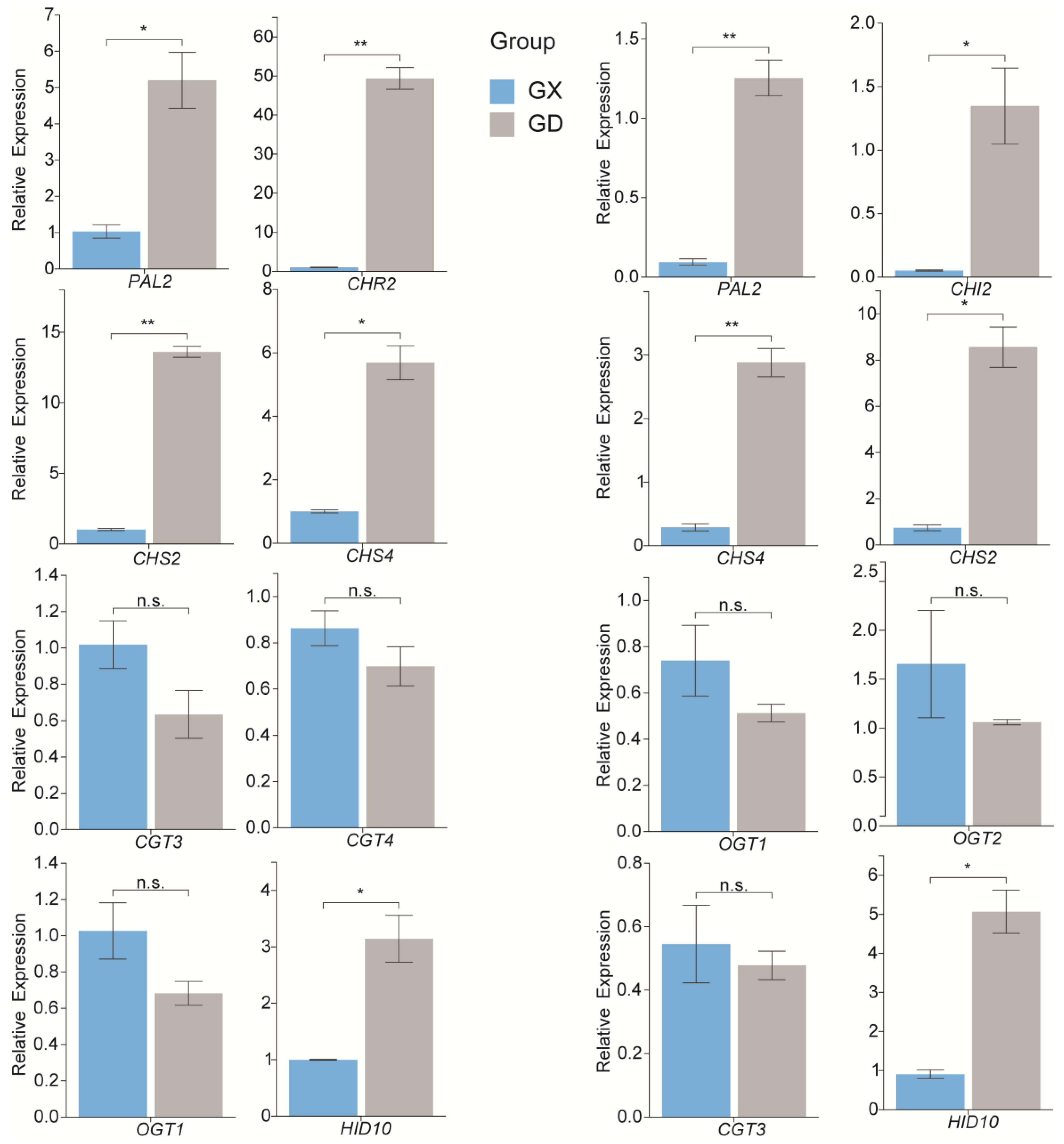
3.7. Quantitative Real-Time Polymerase Chain Reaction Validation of the Expression Levels of the Genes Associated with Puerarin Biosynthesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mo, C.; Wu, Z.; Shang, X.; Shi, P.; Wei, M.; Wang, H.; Xiao, L.; Cao, S.; Lu, L.; Zeng, W.; et al. Chromosome-level and graphic genomes provide insights into metabolism of bioactive metabolites and cold-adaption of Pueraria lobata var. montana. DNA Res. 2022, 29, 1–11. [Google Scholar] [CrossRef]
- Pan, B.; Liu, B.; Yu, Z.-X.; Yang, Y.-Q. Pueraria grandiflora (Fabaceae), a new species from Southwest China. Phytotaxa 2015, 203, 287–291. [Google Scholar] [CrossRef]
- Zhao, Z.; Guo, P.; Brand, E. A concise classification of bencao (Materia medica). Chin. Med. 2018, 13, 18. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, S.; Wang, S.; Gao, P.; Dai, L. A comprehensive review on Pueraria: Insights on its chemistry and medicinal value. Biomed. Pharmacother. 2020, 131, 110734. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Li, Y.; Qiao, X.; Qian, Y.; Ye, M. Chemistry of the Chinese herbal medicine Puerariae Radix (Ge-Gen): A review (Article). J. Chin. Pharm. Sci. 2014, 23, 347–360. [Google Scholar] [CrossRef]
- Wong, K.H.; Li, G.Q.; Li, K.M.; Razmovski-Naumovski, V.; Chan, K. Kudzu root: Traditional uses and potential medicinal benefits in diabetes and cardiovascular diseases. J. Ethnopharmacol. 2011, 134, 584–607. [Google Scholar] [CrossRef] [PubMed]
- Thapa, P.; Kim, H.M.; Hong, J.-P.; Kim, R.; Paudel, S.B.; Choi, H.; Jang, D.S.; Nam, J.-W. Absolute Quantification of Isoflavones in the Flowers of Pueraria lobata by qHNMR. Plants 2022, 11, 548. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, J.; Wang, F.; Zhang, K.; Guo, X.; Tang, T.; Mu, S.; You, J.; Guo, J. Transcriptomic and metabolomic analyses provide new insights into the appropriate harvest period in regenerated bulbs of Fritillaria hupehensis. Front. Plant Sci. 2023, 14, 1132936. [Google Scholar] [CrossRef]
- He, M.; Yao, Y.; Li, Y.; Yang, M.; Li, Y.; Wu, B.; Yu, D. Comprehensive transcriptome analysis reveals genes potentially involved in isoflavone biosynthesis in Pueraria thomsonii Benth. PLoS ONE 2019, 14, e0217593. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Y.; Zhou, L.; You, S.; Deng, H.; Chen, Y.; Alseekh, S.; Yuan, Y.; Fu, R.; Zhang, Z.; et al. MicroTom Metabolic Network: Rewiring Tomato Metabolic Regulatory Network throughout the Growth Cycle. Mol. Plant 2020, 13, 1203–1218. [Google Scholar] [CrossRef]
- Sun, J.; Qiu, C.; Ding, Y.; Wang, Y.; Sun, L.; Fan, K.; Gai, Z.; Dong, G.; Wang, J.; Li, X.; et al. Fulvic acid ameliorates drought stress-induced damage in tea plants by regulating the ascorbate metabolism and flavonoids biosynthesis. BMC Genom. 2020, 21, 411. [Google Scholar] [CrossRef]
- Yue, X.; Ju, Y.; Zhang, H.; Wang, Z.; Xu, H.; Zhang, Z. Integrated transcriptomic and metabolomic analysis reveals the changes in monoterpene compounds during the development of Muscat Hamburg (Vitis vinifera L.) grape berries. Food Res. Int. 2022, 162, 112065. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.-M.; Cao, L.; Jin, X.; Zhang, Y.; Zhang, M.; Wang, Y. Transcriptomic and metabolomic profiling of melatonin treated soybean (Glycine max L.) under drought stress during grain filling period through regulation of secondary metabolite biosynthesis pathways. PLoS ONE 2020, 15, 0239701. [Google Scholar] [CrossRef]
- Yang, W.; Li, N.; Fan, Y.; Dong, B.; Song, Z.; Cao, H.; Du, T.; Liu, T.; Qi, M.; Niu, L.; et al. Transcriptome analysis reveals abscisic acid enhancing drought resistance by regulating genes related to flavonoid metabolism in pigeon pea. Environ. Exp. Bot. 2021, 191, 104627. [Google Scholar] [CrossRef]
- Akashi, T.; Aoki, T.; Ayabe, S. Molecular and biochemical characterization of 2-hydroxyisoflavanone dehydratase. Involvement of carboxylesterase-like proteins in leguminous isoflavone biosynthesis. Plant Physiol. 2005, 137, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Kochs, G.; Grisebach, H. Enzymic synthesis of isoflavones. Eur. J. Biochem. 1986, 155, 311–318. [Google Scholar] [CrossRef]
- Burbulis, I.E.; Winkel-Shirley, B. Interactions among enzymes of the Arabidopsis flavonoid biosynthetic pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 12929–12934. [Google Scholar] [CrossRef]
- Jung, W.; Yu, O.; Lau, S.M.; O’Keefe, D.P.; Odell, J.; Fader, G.; McGonigle, B. Identification and expression of isoflavone synthase, the key enzyme for biosynthesis of isoflavones in legumes. Nat. Biotechnol. 2000, 18, 208–212. [Google Scholar] [CrossRef]
- Han, R.; Takahashi, H.; Nakamura, M.; Yoshimoto, N.; Suzuki, H.; Shibata, D.; Yamazaki, M.; Saito, K. Transcriptomic landscape of Pueraria lobata demonstrates potential for phytochemical study. Front. Plant Sci. 2015, 6, 00426. [Google Scholar] [CrossRef]
- He, X.; Blount, J.W.; Ge, S.; Tang, Y.; Dixon, R.A. A genomic approach to isoflavone biosynthesis in kudzu (Pueraria lobata). Planta 2011, 233, 843–855. [Google Scholar] [CrossRef]
- Shang, X.; Yi, X.; Xiao, L.; Zhang, Y.; Huang, D.; Xia, Z.; Ou, K.; Ming, R.; Zeng, W.; Wu, D.; et al. Chromosomal-level genome and multi-omics dataset of Pueraria lobata var. thomsonii provide new insights into legume family and the isoflavone and puerarin biosynthesis pathways. Hortic. Res. 2022, 9, uhab035. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, C.; Zhou, C.; Li, J.; Zhang, Y. Molecular characterization of the C-glucosylation for puerarin biosynthesis in Pueraria lobata. Plant J. 2017, 90, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Adolfo, L.M.; Burks, D.; Rao, X.; Alvarez-Hernandez, A.; Dixon, R.A. Evaluation of pathways to the C-glycosyl isoflavone puerarin in roots of kudzu (Pueraria montana lobata). Plant Direct 2022, 6, e442. [Google Scholar] [CrossRef]
- Xi, H.; Zhu, Y.; Sun, W.; Tang, N.; Xu, Z.; Shang, X.; Zhang, Y.; Yan, H.; Li, C. Comparative Transcriptome Analysis of Pueraria lobata Provides Candidate Genes Involved in Puerarin Biosynthesis and Its Regulation. Biomolecules 2023, 13, 170. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kong, D.; Fu, Y.; Sussman, M.R.; Wu, H. The effect of developmental and environmental factors on secondary metabolites in medicinal plants. Plant Physiol. Biochem. 2020, 148, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Committee, T.P. Pharmacopoeia of the People’s Republic of China 2020 Edition; China Medical Science Press: Beijing, China, 2020. [Google Scholar]
- Fraga, C.G.; Clowers, B.H.; Moore, R.J.; Zink, E.M. Signature-Discovery Approach for Sample Matching of a Nerve-Agent Precursor Using Liquid Chromatography−Mass Spectrometry, XCMS, and Chemometrics. Anal. Chem. 2010, 82, 4165–4173. [Google Scholar] [CrossRef]
- Campbell, J.A.; Davies, G.J.; Bulone, V.; Henrissat, B. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem. J. 1997, 326, 929–939. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Jeon, J.E.; Kim, J.G.; Fischer, C.R.; Mehta, N.; Dufour-Schroif, C.; Wemmer, K.; Mudgett, M.B.; Sattely, E. A Pathogen-Responsive Gene Cluster for Highly Modified Fatty Acids in Tomato. Cell 2020, 180, 176–187.e119. [Google Scholar] [CrossRef]
- Li, W.; Wen, L.; Chen, Z.; Zhang, Z.; Pang, X.; Deng, Z.; Liu, T.; Guo, Y. Study on metabolic variation in whole grains of four proso millet varieties reveals metabolites important for antioxidant properties and quality traits. Food Chem. 2021, 357, 129791. [Google Scholar] [CrossRef]
- Meraj, T.A.; Fu, J.; Raza, M.A.; Zhu, C.; Shen, Q.; Xu, D.; Wang, Q. Transcriptional Factors Regulate Plant Stress Responses Through Mediating Secondary Metabolism. Genes 2020, 11, 346. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zeng, W.; Li, C.; Wang, J.; Shang, X.; Xiao, L.; Cao, S.; Zhang, Y.; Xu, S.; Yan, H. Genome-wide identification and expression pattern analysis of R2R3-MYB transcription factor gene family involved in puerarin biosynthesis and response to hormone in Pueraria lobata var. thomsonii. BMC Plant Biol. 2023, 23, 107. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Zhao, H.Y.; Zhang, Q.W.; Li, G.H.; Yang, F.Q.; Wang, Y.; Li, Y.C.; Wang, Y.T. A rapid method for simultaneous determination of 14 phenolic compounds in Radix Puerariae using microwave-assisted extraction and ultra high performance liquid chromatography coupled with diode array detection and time-of-flight mass spectrometry. J. Chromatogr. A 2010, 1217, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Brazier-Hicks, M.; Evans, K.M.; Gershater, M.C.; Puschmann, H.; Steel, P.G.; Edwards, R. The C-glycosylation of flavonoids in cereals. J. Biol. Chem. 2009, 284, 17926–17934. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, A.; Nidetzky, B. Switching betweenO- andC-Glycosyltransferase through Exchange of Active-Site Motifs. Angew. Chem. Int. Ed. 2012, 51, 12879–12883. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Z.; Li, C.; Gou, J.; Zhang, Y. Molecular cloning and characterization of an isoflavone 7-O-glucosyltransferase from Pueraria lobata. Plant Cell Rep. 2014, 33, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- He, J.B.; Zhao, P.; Hu, Z.M.; Liu, S.; Kuang, Y.; Zhang, M.; Li, B.; Yun, C.H.; Qiao, X.; Ye, M. Molecular and Structural Characterization of a Promiscuous C-Glycosyltransferase from Trollius chinensis. Angew. Chem. Int. Ed. 2019, 58, 11513–11520. [Google Scholar] [CrossRef]
- Cao, K.; Wang, B.; Fang, W.; Zhu, G.; Chen, C.; Wang, X.; Li, Y.; Wu, J.; Tang, T.; Fei, Z.; et al. Combined nature and human selections reshaped peach fruit metabolome. Genome Biol. 2022, 23, 146. [Google Scholar] [CrossRef]
- Suntichaikamolkul, N.; Tantisuwanichkul, K.; Prombutara, P.; Kobtrakul, K.; Zumsteg, J.; Wannachart, S.; Schaller, H.; Yamazaki, M.; Saito, K.; De-eknamkul, W.; et al. Transcriptome analysis of Pueraria candollei var. mirifica for gene discovery in the biosyntheses of isoflavones and miroestrol. BMC Plant Biol. 2019, 19, 581. [Google Scholar] [CrossRef]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Describe | Excavation Time |
|---|---|---|
| GX1 | One-year-old root tubers | September 2022 |
| GX2 | Two-year-old root tubers | September 2022 |
| GD1 | One-year-old root tubers | January 2023 |
| GD2 | Two-year-old root tubers | January 2023 |
| Sample | Describe | Excavation Time | Puerarin Content (g/Mean ± SD) |
|---|---|---|---|
| GX1 | One-year-old root tubers | September 2022 | 0.44 ± 0.05 |
| GX2 | Two-year-old root tubers | September 2022 | 0.96 ± 0.11 |
| GD1 | One-year-old root tubers | January 2023 | 0.06 ± 0.00 |
| GD2 | Two-year-old root tubers | January 2023 | 0.38 ± 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, X.; Zhu, T.; Min, X.; He, J.; Hou, C.; Liu, X. Integrated Metabolomic and Transcriptomic Analysis of Puerarin Biosynthesis in Pueraria montana var. thomsonii at Different Growth Stages. Genes 2023, 14, 2230. https://doi.org/10.3390/genes14122230
Hu X, Zhu T, Min X, He J, Hou C, Liu X. Integrated Metabolomic and Transcriptomic Analysis of Puerarin Biosynthesis in Pueraria montana var. thomsonii at Different Growth Stages. Genes. 2023; 14(12):2230. https://doi.org/10.3390/genes14122230
Chicago/Turabian StyleHu, Xinyi, Ting Zhu, Xinyi Min, Jianing He, Cong Hou, and Xia Liu. 2023. "Integrated Metabolomic and Transcriptomic Analysis of Puerarin Biosynthesis in Pueraria montana var. thomsonii at Different Growth Stages" Genes 14, no. 12: 2230. https://doi.org/10.3390/genes14122230
APA StyleHu, X., Zhu, T., Min, X., He, J., Hou, C., & Liu, X. (2023). Integrated Metabolomic and Transcriptomic Analysis of Puerarin Biosynthesis in Pueraria montana var. thomsonii at Different Growth Stages. Genes, 14(12), 2230. https://doi.org/10.3390/genes14122230