
Advances in Vertebrate (Cyto)Genomics Shed New Light on Fish Compositional Genome Evolution



Abstract
1. Introduction
2. Results
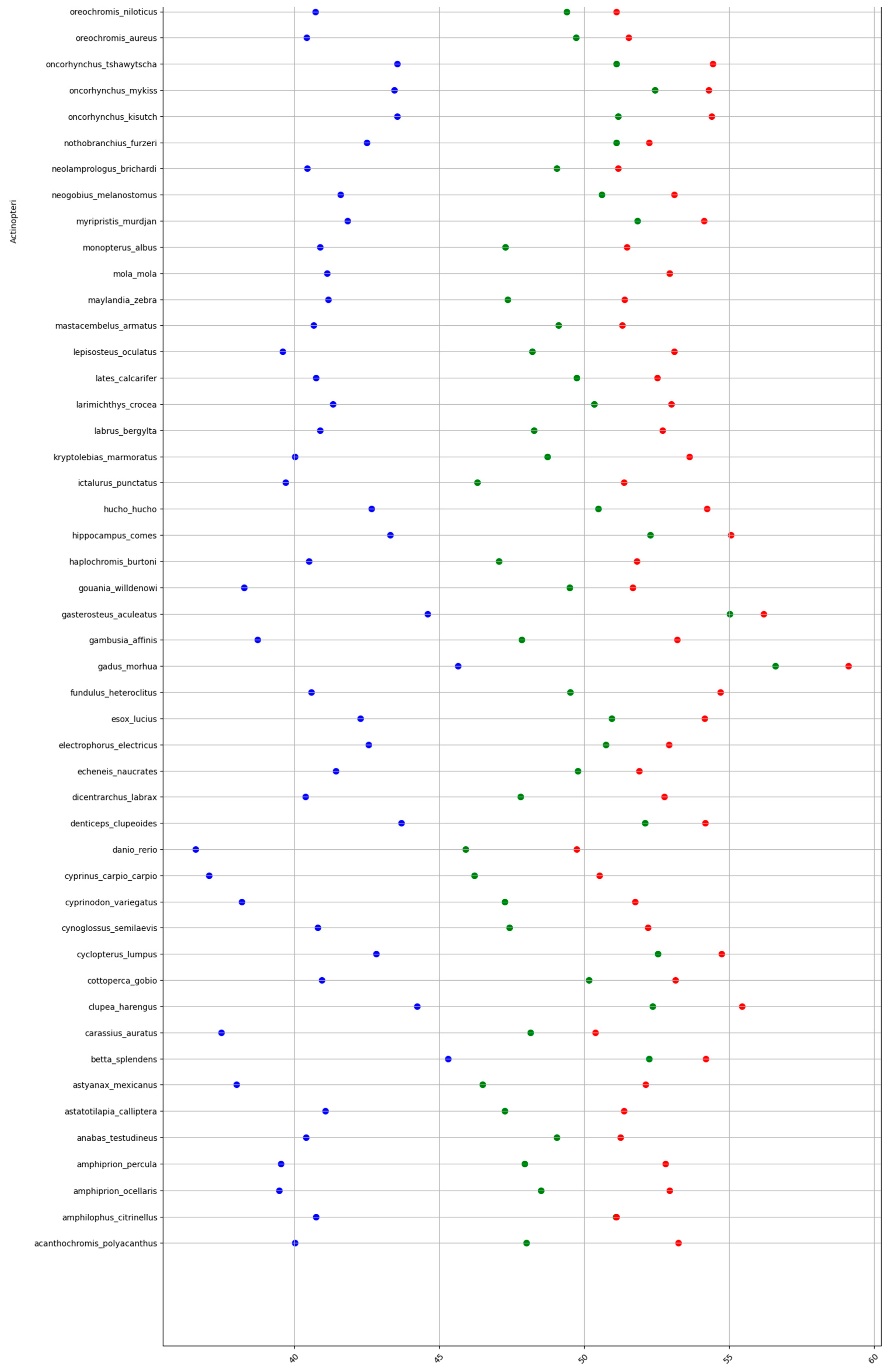
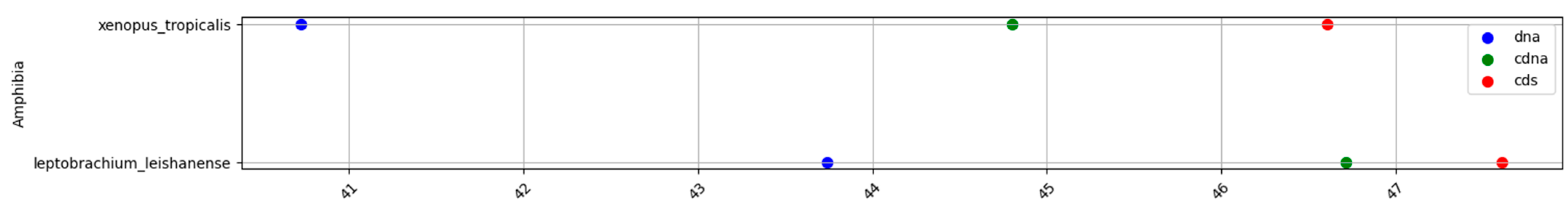
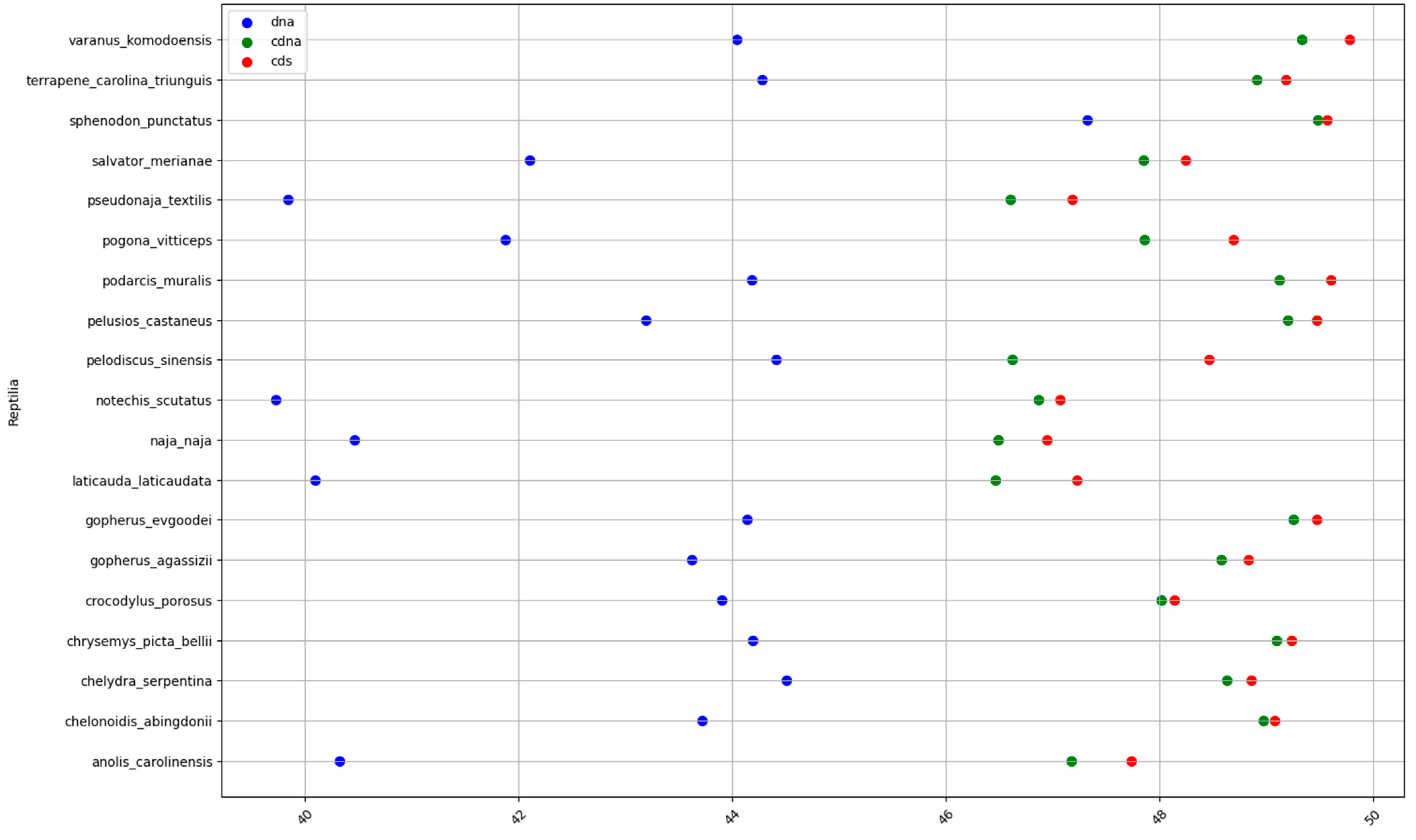
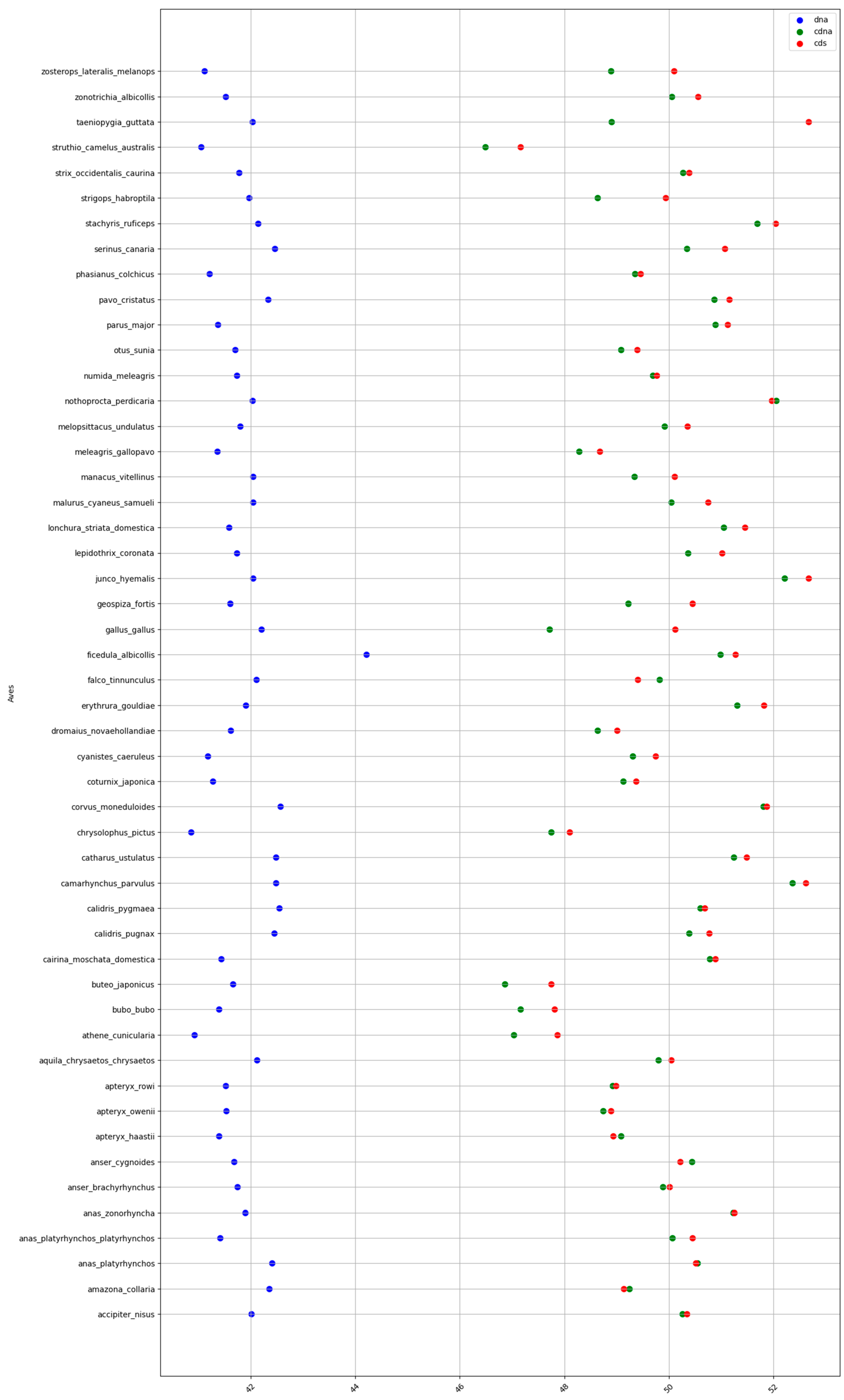
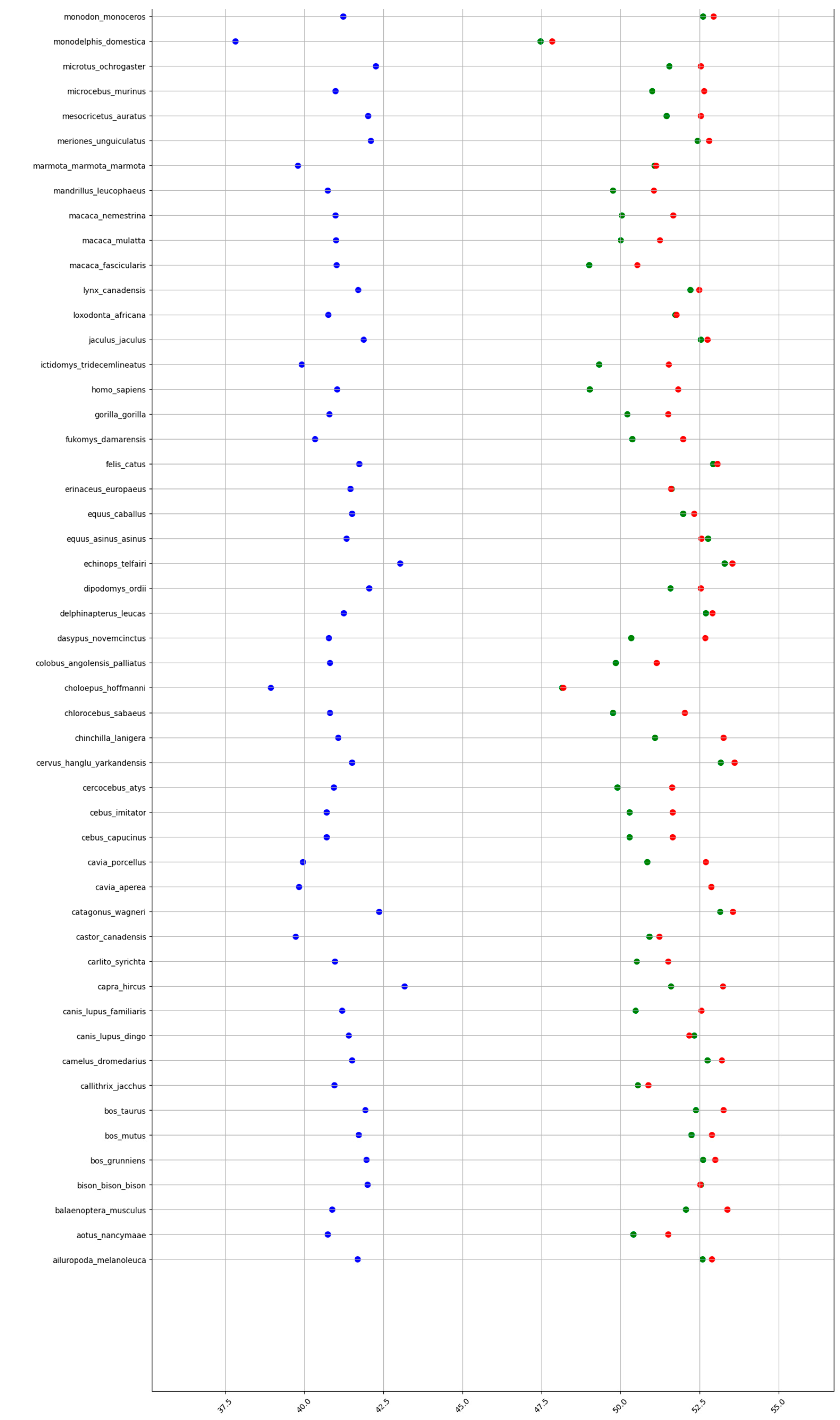
2.1. GC content of the Fractions DNA, cDNA, and cds in Animal Genomes
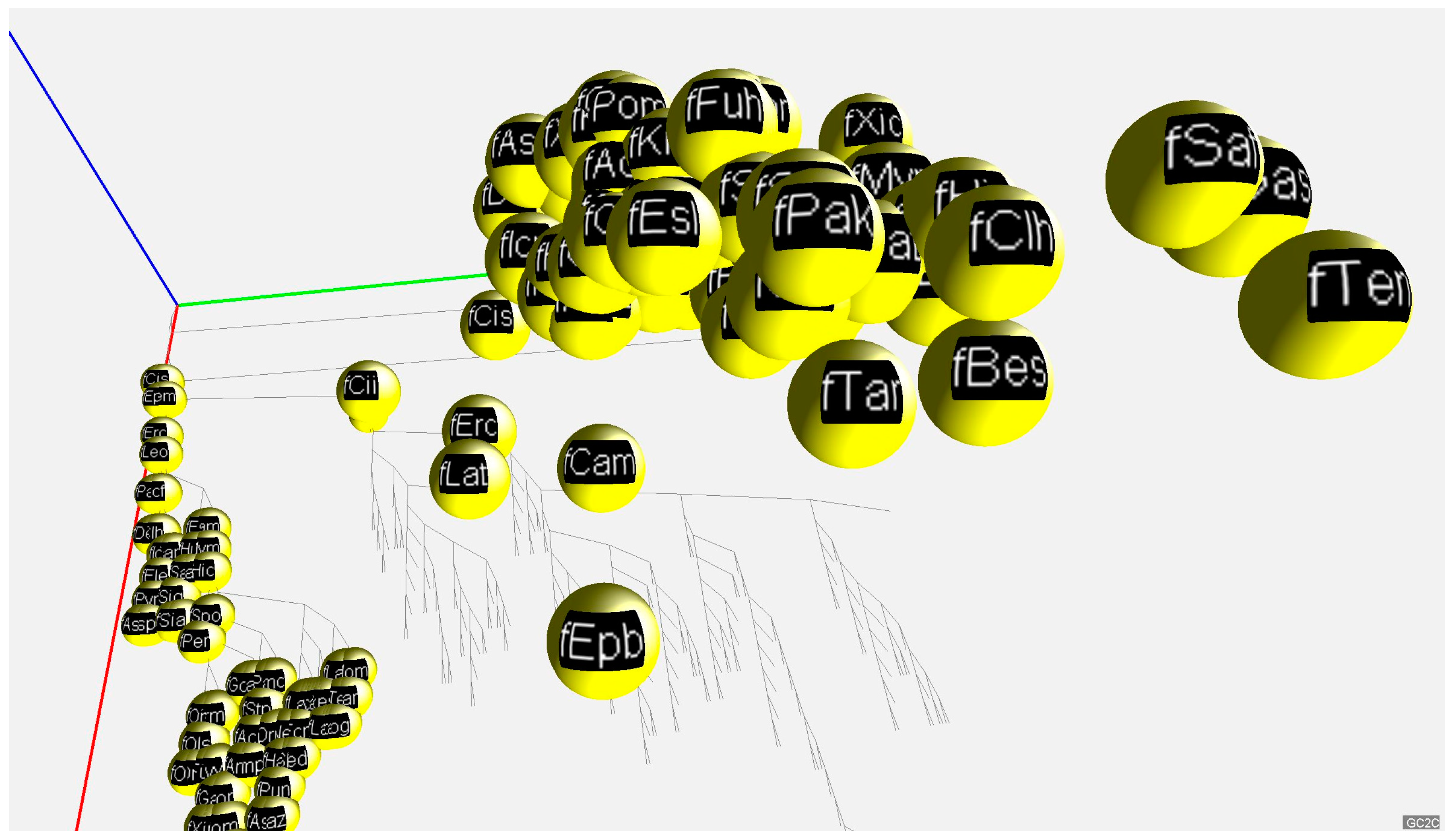
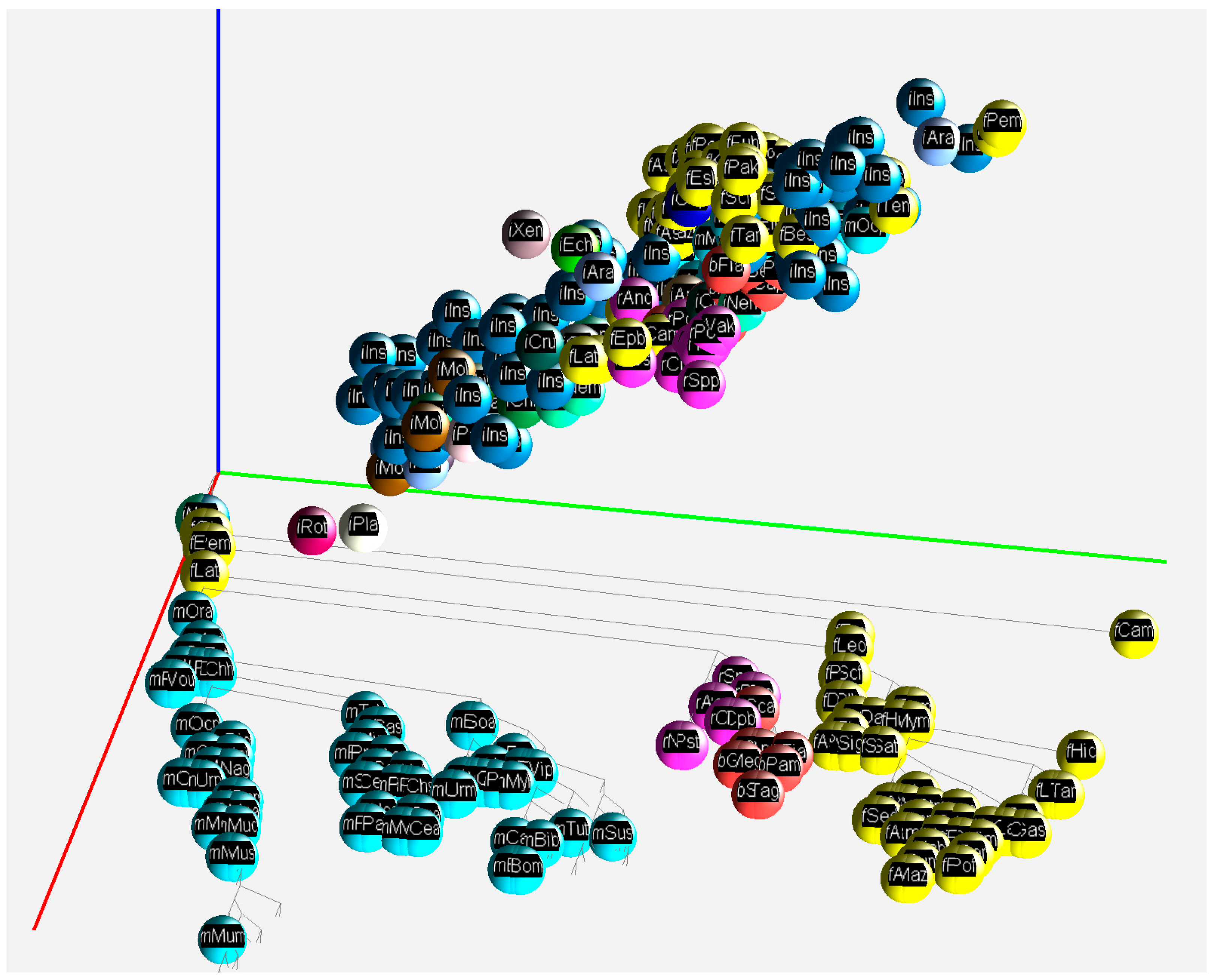
2.2. 3D Visualization of GC% of DNA, cDNA and cds in Animal Genomes
3. Discussion
3.1. Animal Genome Composition and Outlines of Its Evolution
3.2. Quality of Available Genome Assemblies Determines Our Possibilities to Analyse Data
3.3. The Importance of Animal Genome Compositional Data
3.4. Higher Constrains in cDNA and cds GC% in Higher Vertebrates
3.5. Delimiting Genome GC% Values of Invertebrates, Chordates and Higher Vertebrates
4. Materials and Methods
4.1. Data Acquisition and Processing
4.2. Data Treatment and Structure
4.3. 3D Visualization of GC% Data
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Specific Details on Proportions of cDNA and cds in Animal Genomes





References
- Costantini, M.; Cammarano, R.; Bernardi, G. The Evolution of Isochore Patterns in Vertebrate Genomes. BMC Genom. 2009, 10, 146. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, G. The Neoselectionist Theory of Genome Evolution. Proc. Natl. Acad. Sci. USA 2007, 104, 8385–8390. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, G. The Vertebrate Genome: Isochores and Evolution. Mol. Biol. Evol. 1993, 10, 186–204. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Q. Comparative Analysis of the Base Compositions of the Pre-MRNA 3′ Cleaved-Off Region and the MRNA 3′ Untranslated Region Relative to the Genomic Base Composition in Animals and Plants. PLoS ONE 2014, 9, e99928. [Google Scholar] [CrossRef]
- Li, X.-Q.; Du, D. Variation, Evolution, and Correlation Analysis of C+G Content and Genome or Chromosome Size in Different Kingdoms and Phyla. PLoS ONE 2014, 9, e88339. [Google Scholar] [CrossRef]
- Wu, Y.; Yuan, H.; Tan, S.; Chen, J.-Q.; Tian, D.; Yang, H. Increased Complexity of Gene Structure and Base Composition in Vertebrates. J. Genet. Genom. 2011, 38, 297–305. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, Y.; Zhang, W.; Yang, S.; Chen, J.-Q.; Tian, D. Patterns of Exon-Intron Architecture Variation of Genes in Eukaryotic Genomes. BMC Genom. 2009, 10, 47. [Google Scholar] [CrossRef]
- Majtánová, Z.; Symonová, R.; Arias-Rodriguez, L.; Sallan, L.; Ráb, P. “Holostei versus Halecostomi” Problem: Insight from Cytogenetics of Ancient Nonteleost Actinopterygian Fish, Bowfin Amia Calva: Molecular cytogenetics of Amia Calva. J. Exp. Zool. Mol. Dev. Evol. 2017, 328, 620–628. [Google Scholar] [CrossRef]
- Symonová, R.; Majtánová, Z.; Arias-Rodriguez, L.; Mořkovský, L.; Kořínková, T.; Cavin, L.; Pokorná, M.J.; Doležálková, M.; Flajšhans, M.; Normandeau, E.; et al. Genome Compositional Organization in Gars Shows More Similarities to Mammals than to Other Ray-Finned Fish: Cytogenomics of gars. J. Exp. Zool. Mol. Dev. Evol. 2017, 328, 607–619. [Google Scholar] [CrossRef]
- NCBI. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 20 December 2022).
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef]
- Galtier, N. Fine-Scale Quantification of GC-Biased Gene Conversion Intensity in Mammals. Peer Community J. 2021, 1, e17. [Google Scholar] [CrossRef]
- Costantini, M.; Auletta, F.; Bernardi, G. Isochore Patterns and Gene Distributions in Fish Genomes. Genomics 2007, 90, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Borůvková, V.; Howell, W.M.; Matoulek, D.; Symonová, R. Quantitative Approach to Fish Cytogenetics in the Context of Vertebrate Genome Evolution. Genes 2021, 12, 312. [Google Scholar] [CrossRef] [PubMed]
- Symonová, R.; Suh, A. Nucleotide Composition of Transposable Elements Likely Contributes to AT/GC Compositional Homogeneity of Teleost Fish Genomes. Mob. DNA 2019, 10, 49. [Google Scholar] [CrossRef] [PubMed]
- Matoulek, D.; Borůvková, V.; Ocalewicz, K.; Symonová, R. GC and Repeats Profiling along Chromosomes—The Future of Fish Compositional Cytogenomics. Genes 2020, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Graur, D. Slaying (Yet Again) the Brain-Eating Zombie Called the “Isochore Theory”: A Segmentation Algorithm Used to “Confirm” the Existence of Isochores Creates “Isochores” Where None Exist. Int. J. Mol. Sci. 2022, 23, 6558. [Google Scholar] [CrossRef] [PubMed]
- Šmarda, P.; Bureš, P.; Horová, L.; Leitch, I.J.; Mucina, L.; Pacini, E.; Tichý, L.; Grulich, V.; Rotreklová, O. Ecological and Evolutionary Significance of Genomic GC Content Diversity in Monocots. Proc. Natl. Acad. Sci. USA 2014, 111, E4096–E4102. [Google Scholar] [CrossRef] [PubMed]
- Trávníček, P.; Čertner, M.; Ponert, J.; Chumová, Z.; Jersáková, J.; Suda, J. Diversity in Genome Size and GC Content Shows Adaptive Potential in Orchids and Is Closely Linked to Partial Endoreplication, Plant Life-history Traits and Climatic Conditions. New Phytol. 2019, 224, 1642–1656. [Google Scholar] [CrossRef]
- Bowers, J.E.; Tang, H.; Burke, J.M.; Paterson, A.H. GC Content of Plant Genes Is Linked to Past Gene Duplications. PLoS ONE 2022, 17, e0261748. [Google Scholar] [CrossRef]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR MRNA Structures in Eukaryotic Translation Regulation and How to Find Them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef]
- Litterman, A.J.; Kageyama, R.; Le Tonqueze, O.; Zhao, W.; Gagnon, J.D.; Goodarzi, H.; Erle, D.J.; Ansel, K.M. A Massively Parallel 3′ UTR Reporter Assay Reveals Relationships between Nucleotide Content, Sequence Conservation, and MRNA Destabilization. Genome Res. 2019, 29, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Gozashti, L.; Roy, S.W.; Thornlow, B.; Kramer, A.; Ares, M.; Corbett-Detig, R. Transposable Elements Drive Intron Gain in Diverse Eukaryotes. Proc. Natl. Acad. Sci. USA 2022, 119, e2209766119. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.F.; Kautt, A.F.; Nater, A.; Härer, A.; Liang, Y.; Henning, F.; Meyer, A. An Intronic Transposon Insertion Associates with a Trans-Species Color Polymorphism in Midas Cichlid Fishes. Nat. Commun. 2022, 13, 296. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten Things You Should Know about Transposable Elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Furuno, M.; Kasukawa, T.; Saito, R.; Adachi, J.; Suzuki, H.; Baldarelli, R.; Hayashizaki, Y.; Okazaki, Y. CDS Annotation in Full-Length CDNA Sequence. Genome Res. 2003, 13, 1478–1487. [Google Scholar] [CrossRef]
- Smith, J.J.; Kuraku, S.; Holt, C.; Sauka-Spengler, T.; Jiang, N.; Campbell, M.S.; Yandell, M.D.; Manousaki, T.; Meyer, A.; Bloom, O.E.; et al. Sequencing of the Sea Lamprey (Petromyzon Marinus) Genome Provides Insights into Vertebrate Evolution. Nat. Genet. 2013, 45, 415–421. [Google Scholar] [CrossRef]
- Smith, J.J.; Timoshevskaya, N.; Ye, C.; Holt, C.; Keinath, M.C.; Parker, H.J.; Cook, M.E.; Hess, J.E.; Narum, S.R.; Lamanna, F.; et al. The Sea Lamprey Germline Genome Provides Insights into Programmed Genome Rearrangement and Vertebrate Evolution. Nat. Genet. 2018, 50, 270–277. [Google Scholar] [CrossRef]
- Miyashita, T.; Coates, M.I.; Farrar, R.; Larson, P.; Manning, P.L.; Wogelius, R.A.; Edwards, N.P.; Anné, J.; Bergmann, U.; Palmer, A.R.; et al. Hagfish from the Cretaceous Tethys Sea and a Reconciliation of the Morphological–Molecular Conflict in Early Vertebrate Phylogeny. Proc. Natl. Acad. Sci. USA 2019, 116, 2146–2151. [Google Scholar] [CrossRef]
- Randhawa, S.S.; Pawar, R. Fish Genomes: Sequencing Trends, Taxonomy and Influence of Taxonomy on Genome Attributes. J. Appl. Ichthyol. 2021, 37, 553–562. [Google Scholar] [CrossRef]
- Lu, G.; Luo, M. Genomes of Major Fishes in World Fisheries and Aquaculture: Status, Application and Perspective. Aquac. Fish. 2020, 5, 163–173. [Google Scholar] [CrossRef]
- Amit, M.; Donyo, M.; Hollander, D.; Goren, A.; Kim, E.; Gelfman, S.; Lev-Maor, G.; Burstein, D.; Schwartz, S.; Postolsky, B.; et al. Differential GC Content between Exons and Introns Establishes Distinct Strategies of Splice-Site Recognition. Cell Rep. 2012, 1, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, S. On the Base Composition of Transposable Elements. Int. J. Mol. Sci. 2022, 23, 4755. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Long Road to Long-Read Assembly. Nat. Methods 2021, 18, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Peona, V.; Weissensteiner, M.H.; Suh, A. How Complete Are “Complete” Genome Assemblies?-An Avian Perspective. Mol. Ecol. Res. 2018, 18, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The Complete Sequence of a Human Genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Rhie, A.; McCarthy, S.A.; Fedrigo, O.; Damas, J.; Formenti, G.; Koren, S.; Uliano-Silva, M.; Chow, W.; Fungtammasan, A.; Kim, J.; et al. Towards Complete and Error-Free Genome Assemblies of All Vertebrate Species. Nature 2021, 592, 737–746. [Google Scholar] [CrossRef]
- Hon, T.; Mars, K.; Young, G.; Tsai, Y.-C.; Karalius, J.W.; Landolin, J.M.; Maurer, N.; Kudrna, D.; Hardigan, M.A.; Steiner, C.C.; et al. Highly Accurate Long-Read HiFi Sequencing Data for Five Complex Genomes. Sci. Data 2020, 7, 399. [Google Scholar] [CrossRef] [PubMed]
- Francis, W.R.; Wörheide, G. Similar Ratios of Introns to Intergenic Sequence across Animal Genomes. Genome Biol. Evol. 2017, 9, 1582–1598. [Google Scholar] [CrossRef]
- Hertel, K.J. Combinatorial Control of Exon Recognition. J. Biol. Chem. 2008, 283, 1211–1215. [Google Scholar] [CrossRef]
- Georgakopoulos-Soares, I.; Parada, G.E.; Hemberg, M. Secondary Structures in RNA Synthesis, Splicing and Translation. Comput. Struct. Biotechnol. J. 2022, 20, 2871–2884. [Google Scholar] [CrossRef]
- Jakt, L.M.; Dubin, A.; Johansen, S.D. Intron Size Minimisation in Teleosts. BMC Genom. 2022, 23, 628. [Google Scholar] [CrossRef] [PubMed]
- Moss, S.P.; Joyce, D.A.; Humphries, S.; Tindall, K.J.; Lunt, D.H. Comparative Analysis of Teleost Genome Sequences Reveals an Ancient Intron Size Expansion in the Zebrafish Lineage. Genome Biol. Evol. 2011, 3, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Cruveiller, S.; Jabbari, K.; Clay, O.; Bernardi, G. Compositional Gene Landscapes in Vertebrates. Genome Res. 2004, 14, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Elhaik, E.; Landan, G.; Graur, D. Can GC Content at Third-Codon Positions Be Used as a Proxy for Isochore Composition? Mol. Biol. Evol. 2009, 26, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Huttener, R.; Thorrez, L.; in’t Veld, T.; Granvik, M.; Snoeck, L.; Van Lommel, L.; Schuit, F. GC Content of Vertebrate Exome Landscapes Reveal Areas of Accelerated Protein Evolution. BMC Evol. Biol. 2019, 19, 144. [Google Scholar] [CrossRef] [PubMed]
- Braasch, I.; Gehrke, A.R.; Smith, J.J.; Kawasaki, K.; Manousaki, T.; Pasquier, J.; Amores, A.; Desvignes, T.; Batzel, P.; Catchen, J.; et al. The Spotted Gar Genome Illuminates Vertebrate Evolution and Facilitates Human-Teleost Comparisons. Nat. Genet. 2016, 48, 427–437. [Google Scholar] [CrossRef]
- Symonová, R. How (Not) to Read Fish Genomics Data—The Importance of Cytogenomics Knowledge in the Current Flood of Sequenced Genomes. J. Appl. Ichthyol. 2022, 1–4. [Google Scholar] [CrossRef]
- Carotti, E.; Carducci, F.; Canapa, A.; Barucca, M.; Greco, S.; Gerdol, M.; Biscotti, M.A. Transposable Elements and Teleost Migratory Behaviour. Int. J. Mol. Sci. 2021, 22, 602. [Google Scholar] [CrossRef]
- Gaffaroglu, M.; Majtánová, Z.; Symonová, R.; Pelikánová, Š.; Unal, S.; Lajbner, Z.; Ráb, P. Present and Future Salmonid Cytogenetics. Genes 2020, 11, 1462. [Google Scholar] [CrossRef]
- Tarallo, A.; Angelini, C.; Sanges, R.; Yagi, M.; Agnisola, C.; D’Onofrio, G. On the Genome Base Composition of Teleosts: The Effect of Environment and Lifestyle. BMC Genom. 2016, 17, 173. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
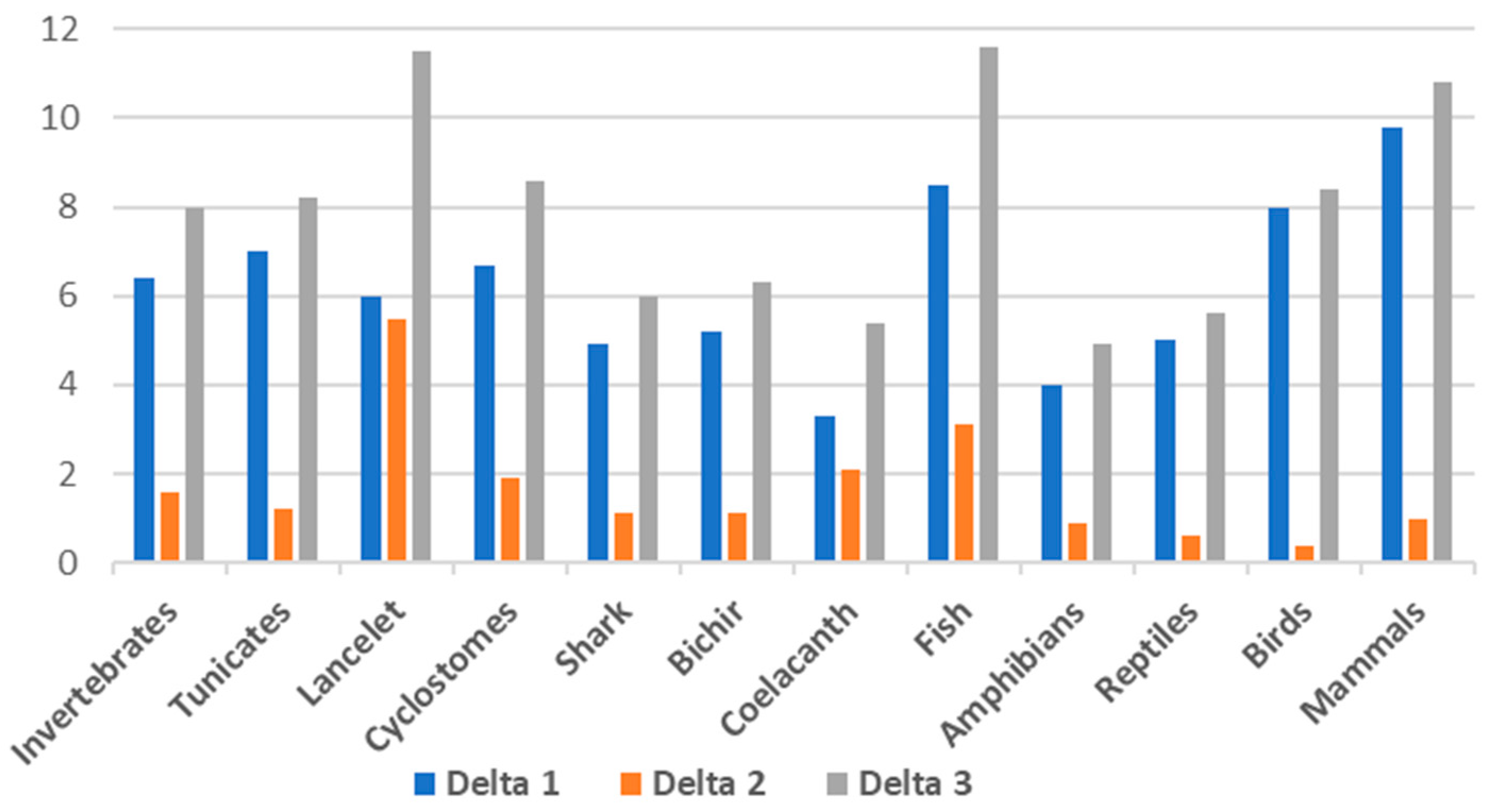
| Group | DNA GC% | cDNA GC% | cds GC% | Delta 1 cDNA-DNA | Delta 2 cds-cDNA | Delta 3 cds-DNA |
|---|---|---|---|---|---|---|
| Invertebrates | 36.1 | 42.5 | 44.1 | 6.4 | 1.6 | 8 |
| Tunicates | 36.0 | 43.0 | 44.2 | 7 | 1.2 | 8.2 |
| Lancelet | 41.5 | 47.5 | 53.0 | 6 | 5.5 | 11.5 |
| Cyclostomes * | 46.3 | 53.0 | 54.9 | 6.7 | 1.9 | 8.6 |
| Shark | 42.3 | 47.2 | 48.3 | 4.9 | 1.1 | 6 |
| Bichir | 39.3 | 44.5 | 45.6 | 5.2 | 1.1 | 6.3 |
| Coelacanth | 41.1 | 44.4 | 46.5 | 3.3 | 2.1 | 5.4 |
| Fish | 40.9 | 49.4 | 52.5 | 8.5 | 3.1 | 11.6 |
| Amphibians | 41.9 | 45.9 | 46.8 | 4 | 0.9 | 4.9 |
| Reptiles | 43.1 | 48.1 | 48.7 | 5 | 0.6 | 5.6 |
| Birds | 41.7 | 49.7 | 50.1 | 8 | 0.4 | 8.4 |
| Mammals | 41.1 | 50.9 | 51.9 | 9.8 | 1 | 10.8 |
| Group | Min DNA GC% | Max DNA GC% |
|---|---|---|
| Invertebrates | Pediculus humanus ~28% | Anopheles gambiae ~44% |
| Cephalochordates | Asymmetron lucayanum ~40% | Branchiostoma floridae ~42% |
| Tetraodontidae ~44–46% | ||
| Fish | Danio rerio ~36% * | Thaleichthys pacificus ~46% |
| Alosa alosa ~48% | ||
| Amphibians | Limnodynastes dumerilii ~37% | Ambystoma mexicanum ~45% |
| Reptiles | Notechis scutatus ~37.2% | Sphaerodactylus townsendi ~46% |
| Birds | Poecile atricapillus ~40% | Pogoniulus pusillus ~46% |
| Mammals | Sarcophilus harrisii ~37–38% Monodelphis domestica ~38% | Ochotona princeps ~44% Ornithorhynchus anatinus ~46% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matoulek, D.; Ježek, B.; Vohnoutová, M.; Symonová, R. Advances in Vertebrate (Cyto)Genomics Shed New Light on Fish Compositional Genome Evolution. Genes 2023, 14, 244. https://doi.org/10.3390/genes14020244
Matoulek D, Ježek B, Vohnoutová M, Symonová R. Advances in Vertebrate (Cyto)Genomics Shed New Light on Fish Compositional Genome Evolution. Genes. 2023; 14(2):244. https://doi.org/10.3390/genes14020244
Chicago/Turabian StyleMatoulek, Dominik, Bruno Ježek, Marta Vohnoutová, and Radka Symonová. 2023. "Advances in Vertebrate (Cyto)Genomics Shed New Light on Fish Compositional Genome Evolution" Genes 14, no. 2: 244. https://doi.org/10.3390/genes14020244
APA StyleMatoulek, D., Ježek, B., Vohnoutová, M., & Symonová, R. (2023). Advances in Vertebrate (Cyto)Genomics Shed New Light on Fish Compositional Genome Evolution. Genes, 14(2), 244. https://doi.org/10.3390/genes14020244