1. Introduction
Small RNAs, such as microRNAs (miRNAs), are important regulators of gene expression. Small interfering RNAs (siRNAs) and small hairpin RNAs (shRNAs) are also widely used tools for biomedicine research and all of these are also currently under development for therapeutic uses in clinics. Although different liposomal carriers and other transfection reagents are useful in the research lab, they are often not sufficient for the in vivo delivery of small RNAs to correct tissue. Over the past decade, cell-originated nanoparticles, extracellular vesicles (EV), have been introduced as a more biocompatible and biological vehicle compared to the other nano-based gene delivery particles. Briefly, in a multicellular organism, intercellular communication and message delivery occur by bilayered lipid vesicles that are naturally released from the cell membrane [
1]. The term EV describes a membrane-bound particle with a diameter of 40 to 1000 nm and subdivisions of EVs such as exosomes (40–100 nm) are termed according to their size and biogenesis [
2]. EV carries genetic cargo (e.g., DNA, mRNA, ncRNA, and proteins) as a part of genetic language during cell-cell crosstalk. Loading of desired nucleic acid into the EVs can be carried out with a variety of methods, either by directly affecting the isolated EVs or modifying the producer cell line to overexpress the desired nucleic acid [
3,
4]. EV engineering provides options for both enhanced cargo loading as well as modifying the EV surface with targeting moieties. Desired nucleic acids, such as miRNAs, can be also enriched into the EVs by a constructed fusion recombinant protein, where a hybrid of EV-packaging domain and miRNA-binding domain is created to enhance the miRNA levels in EVs. Exosome membrane proteins, membrane trafficking proteins or vesicular biogenesis proteins can be applied as a platform for packaging the chosen miRNA binding protein into the exosome [
5,
6]. For example, Li et al. fused the tetraspanin protein CD9 to RNA-binding protein HuR to increase loading of miRNAs into EVs [
7]. The future of EVs as a therapeutic RNA delivery system depends on the development of several factors: firstly, loading of therapeutic RNA should be increased to maximize the levels of active ingredient in the medicine; secondly, targeting the EVs to the target tissue should be enhanced so that the therapeutic RNA can reach the cells where it is needed; and thirdly, internalization of the EVs and the downstream functionality of the therapeutic RNA should be understood in more detail to enhance the proper actions of RNA in the cells.
The purpose of this study was to enhance the loading of miRNAs and similar small RNAs into EVs. Some strategies for loading and modifying the EV-cargo have been identified previously. These include electroporation, freeze-thaw cycles, saponin-mediated loading, and hypotonic dialysis, which have all been studied for use in exogenously loading EVs [
3,
4,
8,
9]. However, these are often not so efficient and, in particular, they are not easy to control and validate for the reliable repetition of experiments. Therefore, engineering the EVs themselves to carry more miRNA is an attractive choice. In this technical study, our preliminary effort was focused on constructing a universal miRNA loading system with high efficiency that could be applied to different producer cell types. Our constructed vector hCD9.hAGO2 expresses an EV membrane recombinant protein related to the exosome biogenesis (CD9 domain) with a universal miRNA-high binding domain (AGO2 domain). To facilitate the ease of use, the fusion recombinant protein was constructed into a lentivirus backbone plasmid with lentiviral particle-producing ability and with a sustained yield promoter. We show that the hCD9.hAGO2 fusion protein significantly enhances miRNA or shRNA packaging into EVs and therefore leads to increased uptake of RNA in the recipient cells.
2. Materials and Methods
2.1. Materials
For generation of the fusion protein, mCLOVER-NLS-AGO2 plasmid (gifted by Dr. Markus Hafner, Laboratory of Muscle Stem Cells and Gene Regulation, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD, USA) was used. Other plasmids used include the backbone pLenti-hPGK plasmid, miR-466c expressing plasmid and shRNA-451 plasmid, which have been published previously [
10,
11].
2.2. Cell Culture
Human embryonic kidney cell line HEK293T (ATCC: CRL-11268), mouse yolk-sac endothelial cell line C166 (ATCC: ATCC CRL-2581) and human retinal pigment epithelial cell line ARPE19 cells (ATCC: CRL-2302) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. HUVECs were cultured in EBM® Medium with 1x EGM BulletKit supplements (Lonza, Germany).
2.3. Amplification of Fragments for hCD9.hAGO2 Fusion Protein Construction
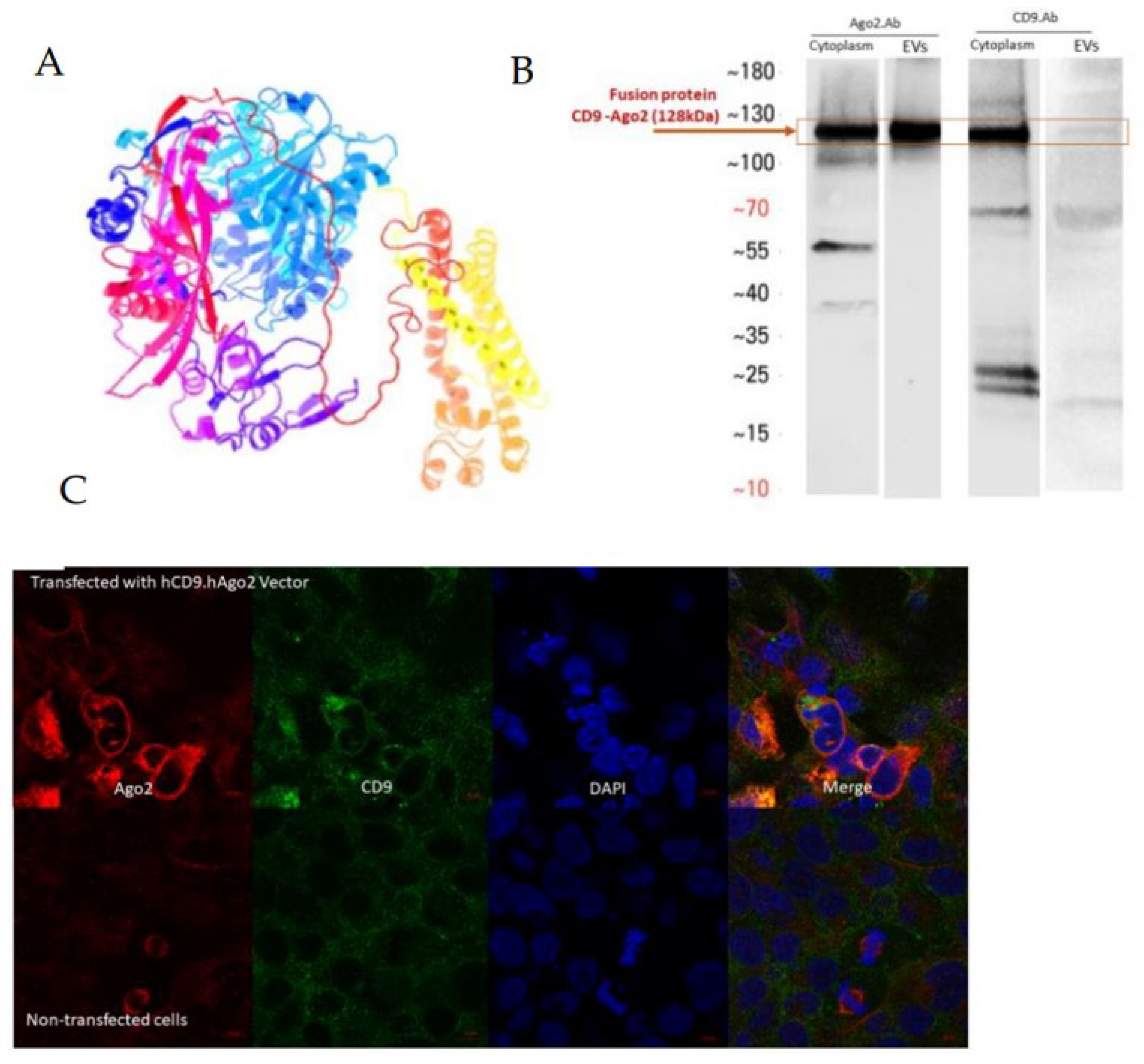
Fusion recombinant protein hCD9.hAGO2 was designed by the SnapGene software (TM1.1.3) and constructed into the pLenti-hPGK backbone (
Figure S1). The protein folding and molecular weights were predicated by the online prediction software Phyre 2 (Protein Homology/analog Y Recognition Engine V 2.0) [
12]. The molecular weight for hCD9.hAGO2 was 129 kDa.
Human AGO2 fragment was amplified from mCLOVER-NLS-AGO2 plasmid, gifted by Dr. Markus Hafner (Laboratory of Muscle Stem Cells and Gene Regulation). The CD9 fragment was amplified from HEK293T cell cDNA.
The cDNA was made by High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems™, Thermo Fisher Scientific, Waltham, MA, USA).
Table 1 shows the In-Fusion primers and touch down PCR programs for amplification of fragments. The CloneAmp HiFi PCR Premix (Takara Bio USA, San Jose, CA, USA) was used for PCR reaction and the In-Fusion
® HD Cloning Kit (Takara Bio USA) was used for fusing and cloning fragments according to the manufacturer’s protocols.
2.4. Transfection
Plasmid transfections were carried out using Polyethyleneimine (PEI, linear, M.W. 25,000; Alfa Aesar, Haverhill, MA, USA) or TransIT-2020 Transfection Reagent (Mirus Bio, Madison, WI, USA), according to the manufacturer’s instructions. siRNA against GAPDH (GAPDH Silencer Pre-designed siRNA, Thermo Fisher) was transfected using TransIT-TKO Transfection Reagent (Mirus Bio), according to the manufacturer’s instructions.
2.5. EV Isolation
Cell culture medium was collected from the cells and pre-cleared by centrifugation (300× g for 5 min, followed with 2000× g for 10 min in +4 °C). CCM was filtered by a 0.2 µm filter and then concentrated using the Amicon® Ultra-15 centrifugal filter device (10 KDa cut-off, Merck Millipore, Burlington, MA, USA). EVs were isolated with qEV Original Size Exclusion Chromatography (SEC) Columns (Izon Science, Lyon, France), according to the manufacturer’s protocol. Fractions 2–4 were pooled as an EV sample unless otherwise stated.
2.6. Western Blot
The vector expression and presence of recombinant protein in the cells and secreted EVs was confirmed by Western blotting.
2.6.1. Sample Preparation for Cells
HEK293T cells were transiently transfected by constructed vectors using PEI. Then, 48 h after transfection, the condition media were removed and cells were frozen. Frozen cell pellets were lysed with 300 µL RIPA lysis buffer (100 mM Tris-base, 2% triton -X100, 1% sodium deoxycholate, 0.2% sodium dodecyl sulfate, 300 mM NaCl, pH 7.4) and incubated 20 min on ice. During ice incubation, samples were vortexed and sonicated three times for 10 s (Branson 2510 Ultrasonic Cleaner). The supernatants (protein) were collected by centrifugation at 14,000× g for 20 min in 4 °C. Then, 48 µL of Lysates were boiled in 12 µL of sample buffer (5X) at 95 °C for 5 min and incubated on ice for SDS page.
2.6.2. Sample Preparation for EVs
For validation of fusion protein packaging in the EVs, 200 µL purified EVs was precipitated by adding 200 µL of water, 400 µL of methanol and 100 µL of chloroform. The sample solution was vortexed and centrifuged 14,000× g for 5 min at room temperature (RT). After discarding the top aqueous layer, the organic phase was washed with 400 µL of methanol and was centrifuged again for protein precipitation. The pellet was air-dried, resuspended and boiled in 30 µL sample buffer(1X) at 95 °C for 5 min. The boiled samples were incubated on ice for SDS page.
2.6.3. SDS-PAGE and Transfer
Next, 28 µL of boiled sample was loaded on 4–15% Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad Laboratories, Hercules, CA, USA) and proteins were transferred onto the PVDF membrane (Trans-Blot Turbo Mini PVDF Transfer Packs, Bio-Rad Laboratories) by Bio-Rad Trans-Blot Turbo transfer system with default program 8–9 V, 10 A for 30 min.
2.6.4. Antibody Staining and Imaging
The fusion proteins were stained with anti-human CD9 antibody (produced in mouse, 60232-1-lg, Proteintech, Rosemont, IL, USA), or anti-human AGO2 antibody (produced in rabbit, ab226943, Abcam, Boston, MA, USA).
The membrane was incubated with 1:1000 diluted primary antibody overnight at 4 °C and then with 1:500 diluted horseradish peroxidase conjugated goat anti-mouse secondary antibody (7076, Cell Signaling Technology, Danvers, MA, USA) and goat anti-rabbit secondary antibody (7074, Cell Signaling Technology) for 2 h at room temperature. Antibody dilutions were made in Immobilon® Block—CH solution (Chemiluminescent Blocker, Merck Millipore, Darmstadt, Germany). After each incubation step, membranes were washed three times in the PBS containing 0.2% Tween-20 for 5 min. Protein bands were detected using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific™) with ChemiDoc XRS System (Bio-Rad Laboratories).
2.7. Double Immunofluorescence Staining and Confocal Laser Scanning Imaging
The double immunofluorescence staining method was used for tracking fused proteins hCD9 and hAGO2 simultaneously. HEK293T cells were cultured on the coverslip in the 6-well plate and constructed vectors transiently transfected via TransIT-2020 Transfection Reagent. Then, 48 h after transfection, at 70–80% confluency, cells were rinsed by PBS to remove the dead cells and debris and fixed with paraformaldehyde (4% PFA in PBS) for 15 min at RT. Cells were washed three times with PBS. Permeabilization was carried out with 0.25% Triton X-100 for 15 min at RT and cells were rinsed three times for 5 min with PBS containing 0.05% Tween-20 (PBST). Unspecific binding sites were blocked with 10% normal goat serum (NGS; Merck Millipore) in PBST for 30 min with gentle shaking in RT.
The primary and secondary antibodies were diluted In staining buffer (5% NGS in PBST). For the simultaneous double staining of fused proteins, two primary antibodies (primary anti-CD9 and primary anti-AGO2) and two secondary antibodies (Alexa Fluor-555 and Alexa Fluor-488) were diluted in the same tube and mixed. The cells were incubated with primary antibodies for 2 h in RT by gently inverting, then washed three times with PBS for 5 min in RT. For secondary staining, the Alexa Fluor-555 (anti-rabbit, 4413S, Cell Signaling Technology) and Alexa Fluor-488 (Goat anti-mouse, 4408S, Cell Signaling Technology) were used. The cells were incubated with secondary antibodies for 1.5 h by gentle shaking in RT and after three times washing with PBS, the coverslips were transferred to the microscope slide with 50 µL of mounting medium with DAPI (Vector Laboratories, Newark, CA, USA) and were sealed with nail polish.
Confocal laser scanning microscopy was performed using a LSM700 Laser Scanning Confocal Microscope (LSM 700; Carl Zeiss Microscopy GmbH). Microscope configuration was the following: The objective lens; Plan-Apochromat 63X/1.40 oil M27. The sequential scanning with stack mode (0.5 µm for Z-scaling and 20.50 µm for stack size). The excitation: 405 nm (Blue: DAPI), 488 nm (Green: Alexa Fluor-488) and 555 nm (Red: Alexa Fluor-555).
2.8. Nanoparticle Tracking Analysis (NTA)
Particle number (concentration) and size distribution of collected EV samples were analyzed with NTA Nanosight NS300 (Malvern Panalytical) equipped with Blue405 laser and sCMOS camera according to the manufacturer’s instructions. Samples were diluted 200–400-fold with filtered PBS (0.2 um). For each sample, four videos of 40 s were captured at 22–25 °C with the camera level 15. Data were analyzed using detection threshold 5. An analysis was performed by instrument software (Nanosight 3.2 Dev Build 3.2.16)
2.9. Cryogenic Electron Microscopy (Cryo-EM)
HEK293T cells transfected with miR-466c plasmid and either with GFP plasmid (ctrl EVs) or hCD9.hAGO2 fusion protein plasmid (hCD9.hAGO2 EVs) using PEI reagent. EVs were isolated from cell culture media with qEV columns and fractions 2–4 were pooled and concentrated with Amicon MWCO 10 kDa filter units (Merck Millipore). EVs for analysis were in PBS buffer with the concentration of 1.91 × 1010 particles/mL as measured with NTA. EV samples were sent to Helsinki HiLIFE Cryo-EM core for imaging.
2.10. RNA Isolation
For the analysis of RNA loading into EVs and transfer to recipient cells, mmu-miR-466c, shRNA-451 or siRNA against GAPDH were transfected to HEK293T cells together with a fusion protein plasmid hCD9.hAGO2 or GFP plasmid as a control. Cells were seeded in density 4–4.5 × 106 cells in 15 cm plate and co-transfected with 15 µg fusion protein or GFP plasmid and 15 µg miRNA or shRNA expression plasmid with 45 µg PEI reagent. For EV collection, cells were cultured in DMEM supplemented with 2% FBS. Then, 4 h after transfection, the media was changed to a fresh complete media. Following 48 h after transfection, the condition media (17 mL per plate) and EVs were isolated as previously described.
The total RNA from purified EVs was isolated by TRIzol (TRIzol® reagent; Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s instructions. Then, 750 µL TRIzol reagent was used for a maximum of 200 µL EVs sample. To increase the efficiency of RNA precipitation, 1 µL GlycoBlue (Invitrogen™, Thermo Fisher Scientific) was added to the lysis sample and incubated at 80 °C for 5 min. The RNA was precipitated with 500 µL isopropanol and incubated at −20 °C overnight. For cell samples, cells were lysed directly on the plate with TRIzol and RNA isolated according to the manufacturer’s instructions. The RNA pellet was dissolved to RNase free water and used for cDNA synthesis.
2.11. cDNA Synthesis and RT-qPCR
For small RNA analysis, cDNA synthesis was performed using the TaqMan MicroRNA Reverse Transcription Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol and analyzed with qPCR using specific TaqMan assays (mmu-miR-466c-3p ID: 464896_mat; mmu-miR-466c-5p ID: 463771_mat, hsa-miR-223 ID: PN4427975, shRNA-451 sense custom assay ID: CS70LFG; shRNA-451 antisense custom assay ID: CSRR86K; Thermo Fisher Scientific). For gene expression analysis, cDNA was synthesized using RevertAid Reverse Transcriptase (Thermo Fisher Scientific) and random hexamer primer (Thermo Fisher Scientific). cDNA quantification was performed using TaqMan Gene Expression Assays (Vegfa ID: Mm00437306_m1; Gapdh ID: Mm99999915_g1; VEGFA ID: Hs00173626_m1; GAPDH ID: 435265, Thermo Fisher Scientific). Samples were quantified by using Maxima Probe/ROX qPCR Master Mix (2X) (Thermo Fisher Scientific). Thermal cycling was performed using a LightCycler® 480 Instrument II (Roche Diagnostics, Rotkreuz, Switzerland) with the following program: 10 min at 95 °C, followed by 50 cycles of 15 s at 95 °C and 60 s at 60 °C. miRNA RT-qPCR was started with an additional step of 2 min at 50 °C. RT-qPCR data were analyzed using the ΔΔCq method where normalization was available.
2.12. Proteinase K and RNase A Treatments
RNase A and Proteinase K treatments were carried out to remove probable free EVs-RNA and free-EVs RNA binding proteins. The cycle threshold (Ct) values from enzyme-treated EVs were compared to no treatment groups. For this purpose, the same number of engineered exosomes per group was incubated with Proteinase K (Thermo Fisher, EO0491) at 37 °C for 30 min with 100 µg/mL final concentration, followed by incubation with RNase A (Thermo Fisher, EN0531) at 37 °C for 15 min with10 µg/mL final concentration. After RNase treatment, the samples were frozen in liquid nitrogen, quickly incubated for 10 min to inactivate the RNase enzyme and then TRIzol reagent was added to the samples.
2.13. Protein Concentration Analysis
Protein concentration of EV fractions was measured with the Qubit protein assay kit (Q33211; Invitrogen, Thermo Fisher Scientific) according to the manufacturer’s instructions.
2.14. MTT Assay
HUVECs were seeded with a seeding density 0.01 × 106 in each well of a 96-well plate at 37 °C in 5% CO2 atmosphere. The day after seeding, 104 EVs per cell were added to the cells. Then, 48 h after EVs incubation, the cells were used for MTT assay according to the manufacturer’s protocol of the MTT cell viability assay kit (Biotium, Inc., Fremont, CA, USA).
2.15. Statistical Analysis
Two-tailed Student’s t-test was used for statistical analysis when applicable. p-value ≤ 0.05 was considered as significant. All data are presented as means ± SD.
4. Discussion
Efficient RNA loading into EVs is important not only for research purposes but also for therapeutic applications as a medicine carrier. This can be achieved with a variety of techniques, such as physically or chemically opening the EV membrane to load cargo or relying on passive loading. It has been shown that the passive RNAs packing into EVs depends on RNA structure and membrane order, which may cause bias in the RNAs that are loaded [
18]. Currently, there are some components proposed for controlling a selective sorting of RNA. For example, most of the miRNAs that are more prominently identified in EVs have a specific short motif that is recognized by heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNPA2B1) as a result of sumoylation [
19]. Therefore, sumoylation of hnRNPA2B1 leads to specifically binding to miR-198 and packaging into the EVs. MS2 protein is a bacteriophage coat protein dimer that possesses RNA-binding activity. Hung and Leonard demonstrated that modified RNA that included three high-affinity loops to MS2 had higher binding activity to bacteriophage proteins located on the surface of EVs [
20]. Nevertheless, more studies on different active loading mechanisms and tools are still required, so that all kinds of RNAs can be loaded into all types of EVs.
In this study, we aimed to actively aid the sorting of short RNAs, namely miRNAs, into EVs. For this purpose, we fused human AGO2, the effective unit of RISC complex binding functional miRNAs in the cells, into the EV transmembrane domain of human CD9 protein. Our rationale was that miRNAs would bind the hCD9.hAGO2 fusion protein in the producer cell efficiently, as it is overexpressed in cells at high levels and would thus compete with the endogenous binding of miRNAs to the cell’s own unmodified AGO2. The CD9 domain would then aid the sorting of the miRNA-fusion protein-complex to EVs during production in producer cells.
As shown in this paper, this construct indeed efficiently increased the amount of miRNA in the EVs compared to only overexpressing the miRNA in producer cells and relying only on the endogenous miRNA sorting to the EVs. We show here that the miRNAs loaded into EVs with the help of the fusion protein are efficiently transferred to recipient cells. This was tested both with human cell line ARPE19 and mouse cell line C166.
Our technical experiments demonstrated that the transfer of miRNAs and shRNAs was both successful, which we also assumed because of their structural similarities. We have previously published shRNA-451 and miR-466c in the regulation of Vegfa expression [
10,
11]. This regulation occurs through targeting the Vegfa gene promoter instead of the mRNA 3′UTR and leads to activation of the gene expression, rather than the decrease in Vegfa levels as in traditional post-transcriptional gene silencing (PTGS). Therefore, we assessed the functional effects of the shRNA or miRNA with EV delivery but were not able to detect effects in the recipient cells. This may be due to many reasons, some of which may be technical (incorrect timepoint of analysis) or due to the functionality of the fusion protein. We were not able to determine whether the shRNA or miRNA is able to transfer to the cell nucleus, where it should exert its effects on the Vegfa gene promoter. Since siRNAs have a more traditional role in the PTGS, we also tried loading siRNA against the GAPDH housekeeping gene to the EVs, but again were not able to see functional effects in the cell. It is possible that the experimental setup was inaccurate for detecting small changes in the gene expression levels of the housekeeping gene, but it is also possible that the siRNA is not exerting proper actions since it is bound to the fusion protein and possibly will not be able to bind functional RISC in the recipient cell. GAPDH is highly expressed in the cells as a housekeeping gene, which are cellular maintenance genes that regulate basic and ubiquitous cellular functions. Therefore, it is also possible that although we increased the siRNA transfer to the recipient cells, the amount of siRNA molecules is still not sufficient for downregulating the mRNA levels in the cells to the extent that it could be significantly detected by qPCR. Interestingly, cell viability was improved by the hCD9.hAGO2-mediated shRNA-451 or miR-466c delivery. This may imply that either the RNAs are released in the cells and can exert their functions, even though we do not detect significant changes in Vegfa expression levels, or the modified EVs themselves have an otherwise positive influence on the treated cells.
In conclusion, the hCD9.hAGO2 fusion protein constructed for this study shows great potential with enhanced small RNA loading to EVs. More work is still required to analyze and understand the process it undergoes in the recipient cell and how to improve the functionality of the small RNA in the cell after the uptake. As different RNA therapies are emerging, tools for delivering the RNA efficiently to target tissue and cells are required and, as natural, non-immunogenic particles, EVs provide an alluring option for drug delivery. The next steps for enhancing endogenous EV delivery vehicles are to enhance drug loading, directing the EV uptake to the desired cell population and enhancing cargo internalization and functionality within the target cells. The study here presents one possible alternative for enhanced RNA loading with hCD9.hAGO2 fusion protein, providing a tool for delivery vehicle development.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}