
Pseudogene TNXA Variants May Interfere with the Genetic Testing of CAH-X

Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Speiser, P.W.; Dupont, B.; Rubinstein, P.; Piazza, A.; Kastelan, A.; New, M.I. High frequency of nonclassical steroid 21-hydroxylase deficiency. Am. J. Hum. Genet. 1985, 37, 650–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannah-Shmouni, F.; Morissette, R.; Sinaii, N.; Elman, M.; Prezant, T.R.; Chen, W.; Pulver, A.; Merke, D.P. Revisiting the prevalence of nonclassic congenital adrenal hyperplasia in US Ashkenazi Jews and Caucasians. Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 1276–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speiser, P.W.; Arlt, W.; Auchus, R.J.; Baskin, L.S.; Conway, G.S.; Merke, D.P.; Meyer-Bahlburg, H.F.L.; Miller, W.L.; Murad, M.H.; Oberfield, S.E.; et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2018, 103, 4043–4088. [Google Scholar] [CrossRef] [Green Version]
- Burch, G.H.; Gong, Y.; Liu, W.; Dettman, R.W.; Curry, C.J.; Smith, L.; Miller, W.L.; Bristow, J. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat. Genet. 1997, 17, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Schalkwijk, J.; Zweers, M.C.; Steijlen, P.M.; Dean, W.B.; Taylor, G.; van Vlijmen, I.M.; van Haren, B.; Miller, W.L.; Bristow, J. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N. Engl. J. Med. 2001, 345, 1167–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zweers, M.C.; Bristow, J.; Steijlen, P.M.; Dean, W.B.; Hamel, B.C.; Otero, M.; Kucharekova, M.; Boezeman, J.B.; Schalkwijk, J. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am. J. Hum. Genet. 2003, 73, 214–217. [Google Scholar] [CrossRef] [Green Version]
- Merke, D.P.; Chen, W.; Morissette, R.; Xu, Z.; Van Ryzin, C.; Sachdev, V.; Hannoush, H.; Shanbhag, S.M.; Acevedo, A.T.; Nishitani, M.; et al. Tenascin-X haploinsufficiency associated with Ehlers-Danlos syndrome in patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2013, 98, E379–E387. [Google Scholar] [CrossRef] [Green Version]
- Morissette, R.; Chen, W.; Perritt, A.F.; Dreiling, J.L.; Arai, A.E.; Sachdev, V.; Hannoush, H.; Mallappa, A.; Xu, Z.; McDonnell, N.B.; et al. Broadening the Spectrum of Ehlers Danlos Syndrome in Patients With Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2015, 100, E1143–E1152. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Merke, D.P. Tenascin-X, Congenital Adrenal Hyperplasia, and the CAH-X Syndrome. Horm. Res. Paediatr. 2018, 89, 352–361. [Google Scholar] [CrossRef]
- Lao, Q.; Brookner, B.; Merke, D.P. High-Throughput Screening for CYP21A1P-TNXA/TNXB Chimeric Genes Responsible for Ehlers-Danlos Syndrome in Patients with Congenital Adrenal Hyperplasia. J. Mol. Diagn. JMD 2019, 21, 924–931. [Google Scholar] [CrossRef]
- Gao, Y.; Lu, L.; Yu, B.; Mao, J.; Wang, X.; Nie, M.; Wu, X. The Prevalence of the Chimeric TNXA/TNXB Gene and Clinical Symptoms of Ehlers-Danlos Syndrome with 21-Hydroxylase Deficiency. J. Clin. Endocrinol. Metab. 2020, 105, 2288–2299. [Google Scholar] [CrossRef] [PubMed]
- Marino, R.; Garrido, N.P.; Ramirez, P.; Notaristéfano, G.; Moresco, A.; Touzon, M.S.; Vaiani, E.; Finkielstain, G.; Obregón, M.G.; Balbi, V.; et al. Ehlers-Danlos Syndrome: Molecular and Clinical Characterization of TNXA/TNXB Chimeras in Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2021, 106, e2789–e2802. [Google Scholar] [CrossRef] [PubMed]
- Paragliola, R.M.; Perrucci, A.; Foca, L.; Urbani, A.; Concolino, P. Prevalence of CAH-X Syndrome in Italian Patients with Congenital Adrenal Hyperplasia (CAH) Due to 21-Hydroxylase Deficiency. J. Clin. Med. 2022, 11, 3818. [Google Scholar] [CrossRef]
- Baumgartner-Parzer, S.; Witsch-Baumgartner, M.; Hoeppner, W. EMQN best practice guidelines for molecular genetic testing and reporting of 21-hydroxylase deficiency. Eur. J. Hum. Genet. EJHG 2020, 28, 1341–1367. [Google Scholar] [CrossRef] [PubMed]
- Lao, Q.; Merke, D.P. Molecular genetic testing of congenital adrenal hyperplasia due to 21-hydroxylase deficiency should include CAH-X chimeras. Eur. J. Hum. Genet. EJHG 2021, 29, 1047–1048. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner-Parzer, S.; Witsch-Baumgartner, M.; Hoeppner, W. Reply to Lao Q and Merke DP. Eur. J. Hum. Genet. EJHG 2021, 29, 1045–1046. [Google Scholar] [CrossRef]
- Blanchong, C.A.; Zhou, B.; Rupert, K.L.; Chung, E.K.; Jones, K.N.; Sotos, J.F.; Zipf, W.B.; Rennebohm, R.M.; Yung Yu, C. Deficiencies of human complement component C4A and C4B and heterozygosity in length variants of RP-C4-CYP21-TNX (RCCX) modules in caucasians. The load of RCCX genetic diversity on major histocompatibility complex-associated disease. J. Exp. Med. 2000, 191, 2183–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkielstain, G.P.; Chen, W.; Mehta, S.P.; Fujimura, F.K.; Hanna, R.M.; Van Ryzin, C.; McDonnell, N.B.; Merke, D.P. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2011, 96, E161–E172. [Google Scholar] [CrossRef]
- New, M.I.; Abraham, M.; Gonzalez, B.; Dumic, M.; Razzaghy-Azar, M.; Chitayat, D.; Sun, L.; Zaidi, M.; Wilson, R.C.; Yuen, T. Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc. Natl. Acad. Sci. USA 2013, 110, 2611–2616. [Google Scholar] [CrossRef] [Green Version]
- Merke, D.P.; Auchus, R.J. Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency. N. Engl. J. Med. 2020, 383, 1248–1261. [Google Scholar] [CrossRef]
- Concolino, P.; Costella, A. Congenital Adrenal Hyperplasia (CAH) due to 21-Hydroxylase Deficiency: A Comprehensive Focus on 233 Pathogenic Variants of CYP21A2 Gene. Mol. Diagn. Ther. 2018, 22, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-H.; Chang, J.-G.; Tsai, C.-H.; Tsai, F.-J.; Chao, H.-T.; Chung, B.-c. Analysis of the Chimeric CYP21P/CYP21 Gene in Steroid 21-Hydroxylase Deficiency. Clin. Chem. 2000, 46, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xu, Z.; Sullivan, A.; Finkielstain, G.P.; Van Ryzin, C.; Merke, D.P.; McDonnell, N.B. Junction site analysis of chimeric CYP21A1P/CYP21A2 genes in 21-hydroxylase deficiency. Clin. Chem. 2012, 58, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beighton, P.; De Paepe, A.; Steinmann, B.; Tsipouras, P.; Wenstrup, R.J. Ehlers-Danlos syndromes: Revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am. J. Med. Genet. 1998, 77, 31–37. [Google Scholar] [CrossRef]
- Levy, H.P. Hypermobile Ehlers-Danlos Syndrome. In GeneReviews(®); Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Bristow, J.; Tee, M.K.; Gitelman, S.E.; Mellon, S.H.; Miller, W.L. Tenascin-X: A novel extracellular matrix protein encoded by the human XB gene overlapping P450c21B. J. Cell Biol. 1993, 122, 265–278. [Google Scholar] [CrossRef] [Green Version]
- Zweers, M.C.; Dean, W.B.; van Kuppevelt, T.H.; Bristow, J.; Schalkwijk, J. Elastic fiber abnormalities in hypermobility type Ehlers-Danlos syndrome patients with tenascin-X mutations. Clin. Genet. 2005, 67, 330–334. [Google Scholar] [CrossRef]
- Chen, W.; Kim, M.S.; Shanbhag, S.; Arai, A.; VanRyzin, C.; McDonnell, N.B.; Merke, D.P. The phenotypic spectrum of contiguous deletion of CYP21A2 and tenascin XB: Quadricuspid aortic valve and other midline defects. Am. J. Med. Genetics. Part A 2009, 149A, 2803–2808. [Google Scholar] [CrossRef] [Green Version]
- Lao, Q.; Mallappa, A.; Rueda Faucz, F.; Joyal, E.; Veeraraghavan, P.; Chen, W.; Merke, D.P. A TNXB splice donor site variant as a cause of hypermobility type Ehlers-Danlos syndrome in patients with congenital adrenal hyperplasia. Mol. Genet. Genom. Med. 2021, 9, e1556. [Google Scholar] [CrossRef]
- Chen, W.; Perritt, A.F.; Morissette, R.; Dreiling, J.L.; Bohn, M.F.; Mallappa, A.; Xu, Z.; Quezado, M.; Merke, D.P. Ehlers-Danlos Syndrome Caused by Biallelic TNXB Variants in Patients with Congenital Adrenal Hyperplasia. Hum. Mutat. 2016, 37, 893–897. [Google Scholar] [CrossRef] [Green Version]
- Hogan, G.J.; Vysotskaia, V.S.; Beauchamp, K.A.; Seisenberger, S.; Grauman, P.V.; Haas, K.R.; Hong, S.H.; Jeon, D.; Kash, S.; Lai, H.H.; et al. Validation of an Expanded Carrier Screen that Optimizes Sensitivity via Full-Exon Sequencing and Panel-wide Copy Number Variant Identification. Clin. Chem. 2018, 64, 1063–1073. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, W.; Merke, D.P.; McDonnell, N.B. Comprehensive mutation analysis of the CYP21A2 gene: An efficient multistep approach to the molecular diagnosis of congenital adrenal hyperplasia. J. Mol. Diagn. JMD 2013, 15, 745–753. [Google Scholar] [CrossRef] [Green Version]
- Lao, Q.; Jardin, M.D.; Jayakrishnan, R.; Ernst, M.; Merke, D.P. Complement component 4 variations may influence psychopathology risk in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum. Genet. 2018, 137, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Kolli, V.; Kim, H.; Rao, H.; Lao, Q.; Gaynor, A.; Milner, J.D.; Merke, D.P. Measurement of serum tenascin-X in patients with congenital adrenal hyperplasia at risk for Ehlers-Danlos contiguous gene deletion syndrome CAH-X. BMC Res. Notes 2019, 12, 711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keen-Kim, D.; Redman, J.B.; Alanes, R.U.; Eachus, M.M.; Wilson, R.C.; New, M.I.; Nakamoto, J.M.; Fenwick, R.G. Validation and clinical application of a locus-specific polymerase chain reaction- and minisequencing-based assay for congenital adrenal hyperplasia (21-hydroxylase deficiency). J. Mol. Diagn. JMD 2005, 7, 236–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olney, R.C.; Mougey, E.B.; Wang, J.; Shulman, D.I.; Sylvester, J.E. Using Real-Time, Quantitative PCR for Rapid Genotyping of the Steroid 21-Hydroxylase Gene in a North Florida Population. J. Clin. Endocrinol. Metab. 2002, 87, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Cantürk, C.; Baade, U.; Salazar, R.; Storm, N.; Pörtner, R.; Höppner, W. Sequence analysis of CYP21A1P in a German population to aid in the molecular biological diagnosis of congenital adrenal hyperplasia. Clin. Chem. 2011, 57, 511–517. [Google Scholar] [CrossRef]
- Tsai, L.-P.; Cheng, C.-F.; Chuang, S.-H.; Lee, H.-H. Analysis of the CYP21A1P pseudogene: Indication of mutational diversity and CYP21A2-like and duplicated CYP21A2 genes. Anal. Biochem. 2011, 413, 133–141. [Google Scholar] [CrossRef]
- Tsai, L.P.; Lee, H.H. Analysis of CYP21A1P and the duplicated CYP21A2 genes. Gene 2012, 506, 261–262. [Google Scholar] [CrossRef]
- Lee, H.H. Variants of the CYP21A2 and CYP21A1P genes in congenital adrenal hyperplasia. Clin. Chim. Acta Int. J. Clin. Chem. 2013, 418, 37–44. [Google Scholar] [CrossRef]
- Mueller, P.W.; Lyons, J.; Kerr, G.; Haase, C.P.; Isett, R.B. Standard enrichment methods for targeted next-generation sequencing in high-repeat genomic regions. Genet. Med. Off. J. Am. Coll. Med. Genet. 2013, 15, 910–911. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285. [Google Scholar] [CrossRef]
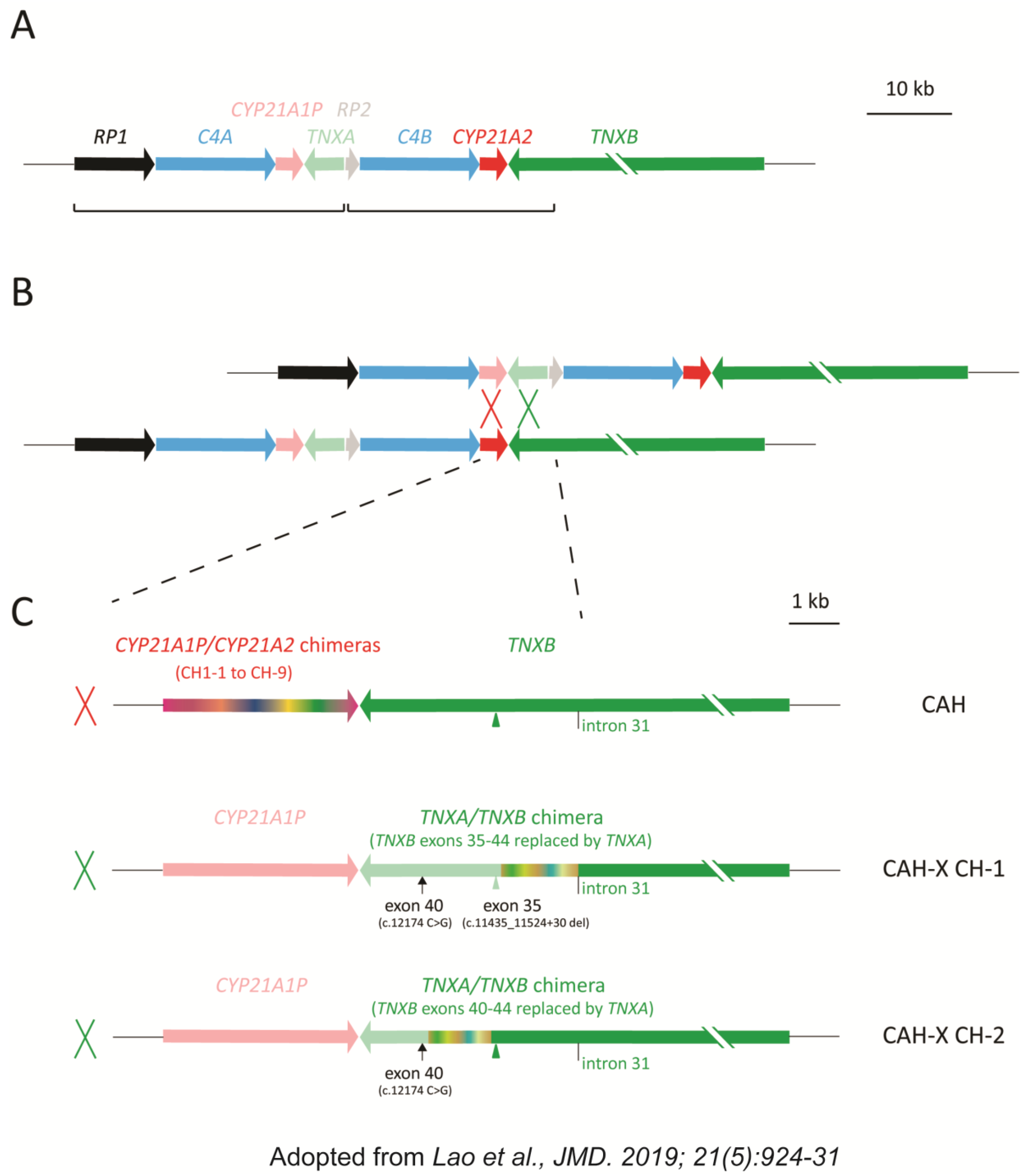
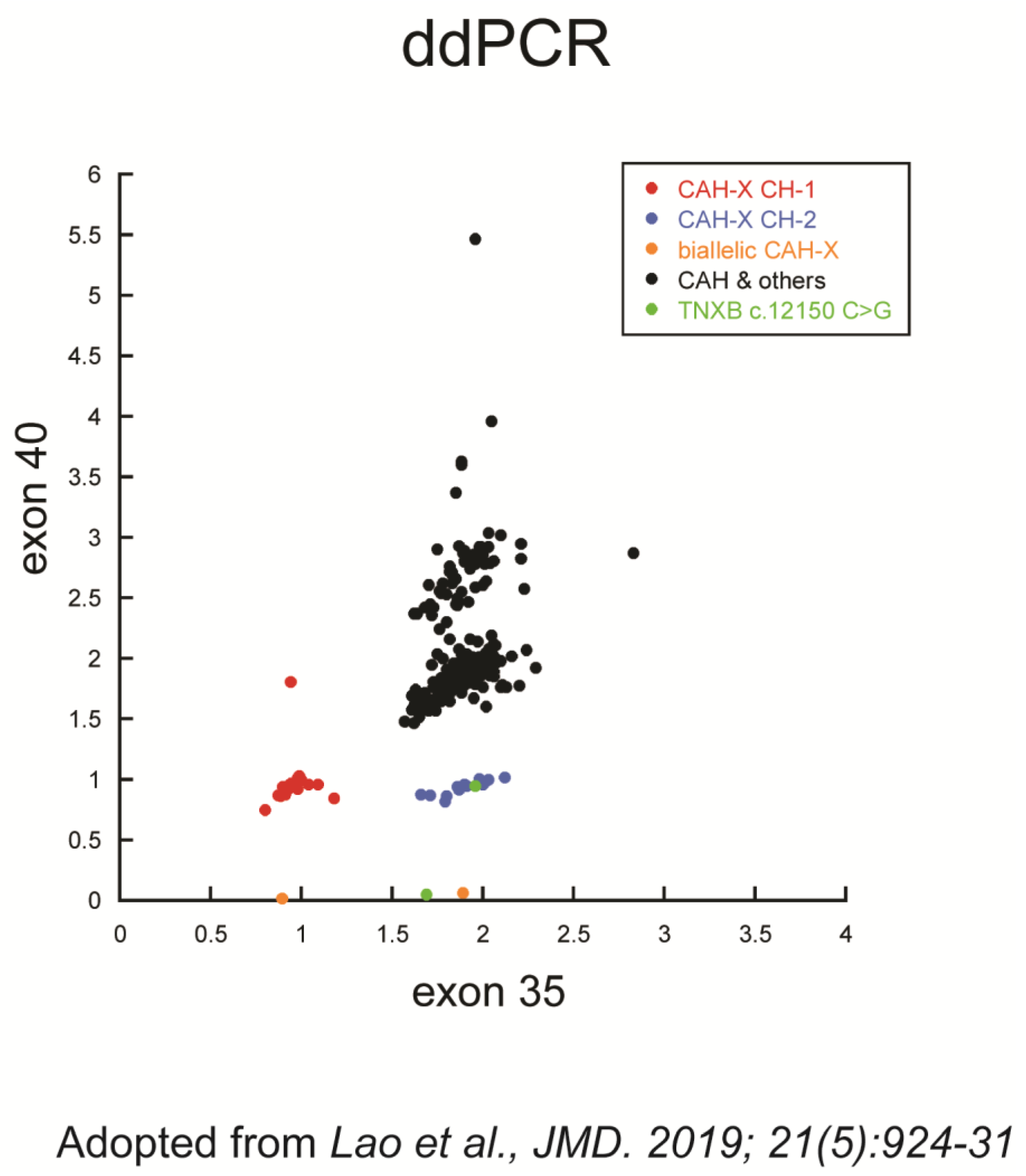

{kind=link}
{kind=link}
| Subject# | CAH 21OHD or Other Disease Status | CYP21A2 Genotype | TNXB X40 CNV (ddPCR) | Total TNXA CNV | TNXA Variant (X40-like) | In cis CYP21A2 Allele |
|---|---|---|---|---|---|---|
| 1 | SW | In2G/In2G | 5.47 | 3 | 3 | In2G/In2G |
| 2 | SW | CAH-CH1/In2G | 2.89 | 1 | 1 | In2G |
| 3 | SW | R356W/In2G | 2.79 | 3 | 1 | N/D |
| 4 | SW | Q318X/CAH-CH5 | 2.62 | 1 | 1 | Q318X |
| 5 | SW | In2G/CAH-CH5 | 2.66 | 1 | 1 | In2G |
| 6 | SW | In2G/In2G | 2.55 | 2 | 1 | In2G |
| 7 | SW | In2G/Q318X | 2.71 | 2 | 1 | N/D |
| 8 | SW | In2G/CAH-CH1 | 2.87 | 1 | 1 | In2G |
| 9 | SW | CAH-CH1/IVS8+1G>A | 2.72 | 1 | 1 | IVS8+1G>A |
| 10 | SV | CAH-CH1/I172N | 2.64 | 1 | 1 | I172N |
| 11 | SV | In2G/In2G | 2.59 | 1 | 1 | In2G |
| 12 | SV | CAH-CH1/del-I172N | 2.81 | 2 | 1 | del-I172N |
| 13 | SV | I172N/In2G | 2.78 | 2 | 1 | N/D |
| 14 | SV | In2G/E6cluster/I172N | 2.5 | 2 | 1 | N/D |
| 15 | SV | I172N/In2G | 2.58 | 2 | 1 | N/D |
| 16 | SV | I172N/Q318X | 2.86 | 1 | 1 | N/D |
| 17 | SV | I172N/Q318X | 2.8 | 2 | 1 | Q318X |
| 12B | NC | del/del-I172N | 2.74 | 2 | 1 | del-I172N |
| 2B | carrier | In2G/normal | 2.61 | 2 | 1 | In2G |
| 4A | carrier | Q318X/normal | 2.9 | 2 | 1 | Q318X |
| 6B | carrier | In2G/normal | 2.76 | 2 | 1 | In2G |
| 8A | carrier | In2G/normal | 2.92 | 2 | 1 | In2G |
| 11A | carrier | In2G/normal | 3.37 | 2 | 2 | In2G/normal |
| 18A | carrier | In2G/normal | 2.78 | 2 | 1 | normal |
| 19A | carrier | CAH-CH1/normal | 2.83 | 1 | 1 | normal |
| 20A | carrier | CAH-CH1/normal | 2.84 | 2 | 1 | normal |
| 21A | carrier | CAH-CH5/normal | 2.55 | 2 | 1 | normal |
| 21C | carrier | P453S/normal | 2.61 | 2 | 1 | normal |
| 22B | carrier | V281L/normal | 2.56 | 3 | 1 | normal |
| 23A | carrier | CAH-CH3/normal | 3.63 | 2 | 2 | normal * |
| 24A | carrier | In2G/normal | 2.69 | 2 | 1 | N/D |
| 25A | carrier | I172N/normal | 2.62 | 1 | 0 | N/A |
| 26B | carrier | CAH-CH5/normal | 2.81 | 1 | 1 | normal |
| 27B | carrier | Q318X/normal | 2.92 | 3 | N/D † | N/A |
| 28B | carrier | V281L/normal | 2.95 | 3 | 1 | normal |
| 29A | carrier | In2G/normal | 2.77 | 2 | 1 ‡ | normal |
| 20C | normal | normal/normal | 2.68 | 2 | 1 | normal |
| 30C | normal | normal/normal | 2.54 | 2 | 1 | normal |
| 31A | normal | normal/normal | 2.93 | 2 | 1 | normal |
| 32 | XY DSD | normal/normal | 3.6 | 2 | 2 | normal/normal |
| 33 | Adrenal insufficiency | normal/normal | 2.87 | 2 | 0 | N/A § |
| 34 | Aldosterone synthase deficiency | normal/normal | 2.81 | 3 | 1 | normal |
| 35A | 11βOHD carrier | normal/normal | 2.92 | 2 | 1 | normal |
| 36 | 17OHD CAH | normal/normal | 3.96 | 3 | 2 | normal/normal ¶ |
| 37A | 11βOHD carrier | normal/normal | 2.86 | 2 | 1 | normal |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lao, Q.; Zhou, K.; Parker, M.; Faucz, F.R.; Merke, D.P. Pseudogene TNXA Variants May Interfere with the Genetic Testing of CAH-X. Genes 2023, 14, 265. https://doi.org/10.3390/genes14020265
Lao Q, Zhou K, Parker M, Faucz FR, Merke DP. Pseudogene TNXA Variants May Interfere with the Genetic Testing of CAH-X. Genes. 2023; 14(2):265. https://doi.org/10.3390/genes14020265
Chicago/Turabian StyleLao, Qizong, Kiet Zhou, Megan Parker, Fabio R. Faucz, and Deborah P. Merke. 2023. "Pseudogene TNXA Variants May Interfere with the Genetic Testing of CAH-X" Genes 14, no. 2: 265. https://doi.org/10.3390/genes14020265