
DNA Double-Strand Break-Related Competitive Endogenous RNA Network of Noncoding RNA in Bovine Cumulus Cells

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. CCs Collection, Culture and BLM Treatment
2.2. γH2AX Detection
2.3. Cell Cycle Analysis
2.4. Cell Proliferation Assays
2.5. Apoptosis Analysis
2.6. RNA Extraction
2.7. LncRNA, circRNA, and mRNA Library Construction
2.8. miRNA Library Construction
2.9. Bioinformatic Analysis of LncRNA
2.10. Bioinformatic Analysis of circRNAs
2.11. Bioinformatic Analysis of miRNA
2.12. Expression Analysis
2.13. Functional Enrichment Analysis
2.14. RT–qPCR
2.15. CeRNA Regulatory Network Analysis
2.16. Statistical Analysis
3. Results
3.1. BLM Treatment Induced DSBs, Disrupted the Cell Cycle, and Inhibited Cell Proliferation in Bovine CCs
3.2. Overview of RNA Sequencing
3.3. Differentially Expressed mRNA Identification
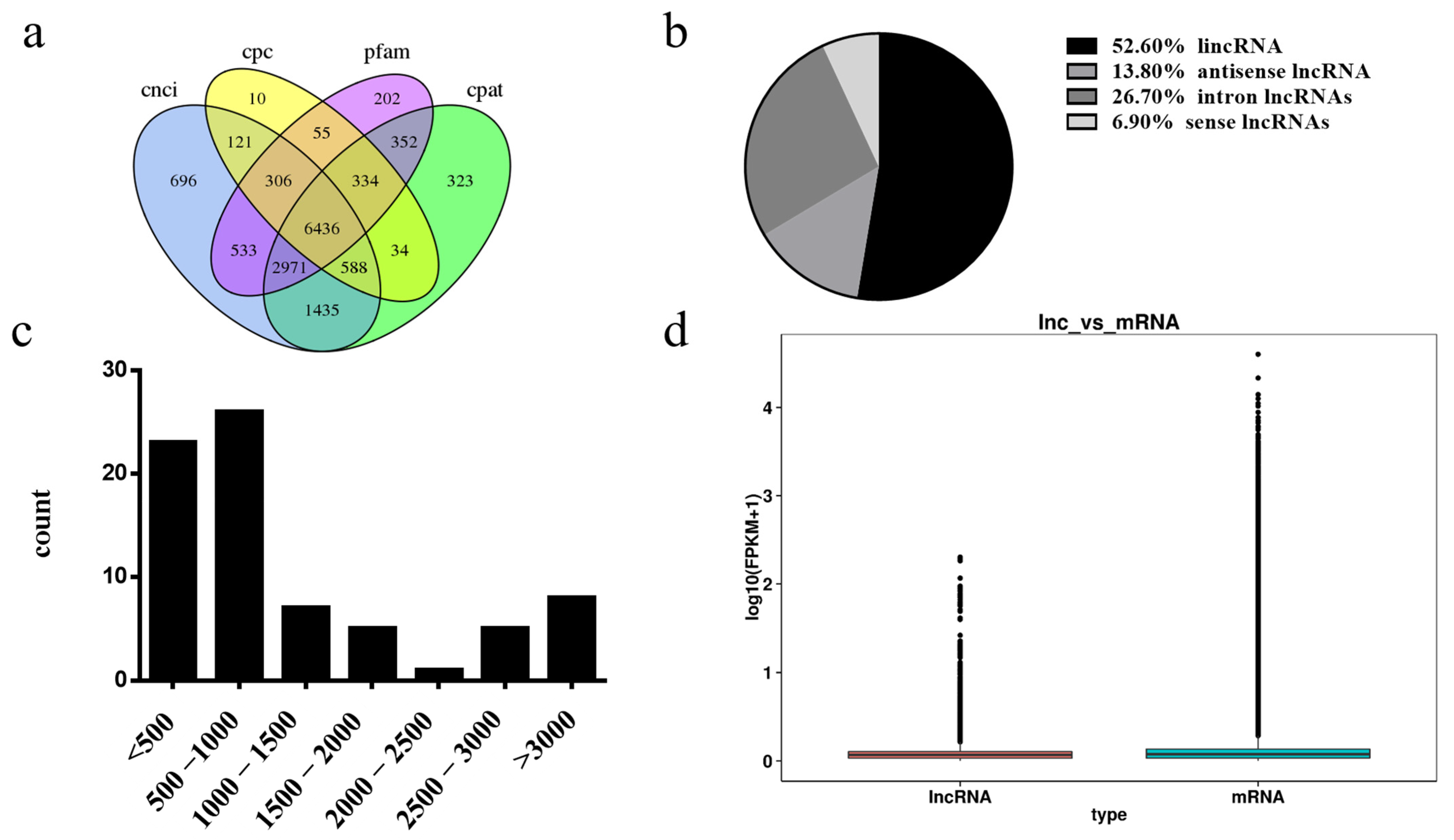
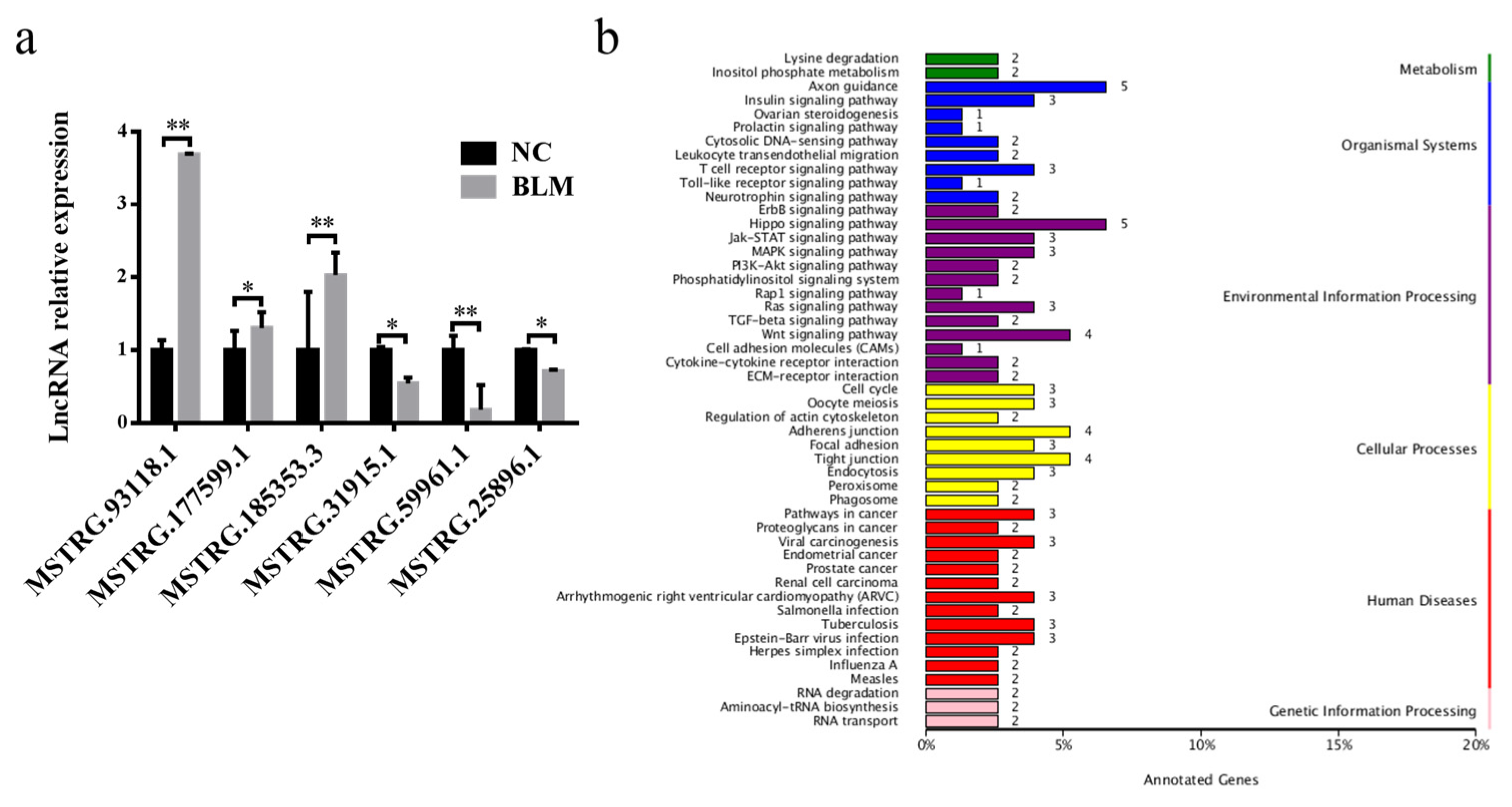
3.4. Differentially Expressed lncRNA Identification
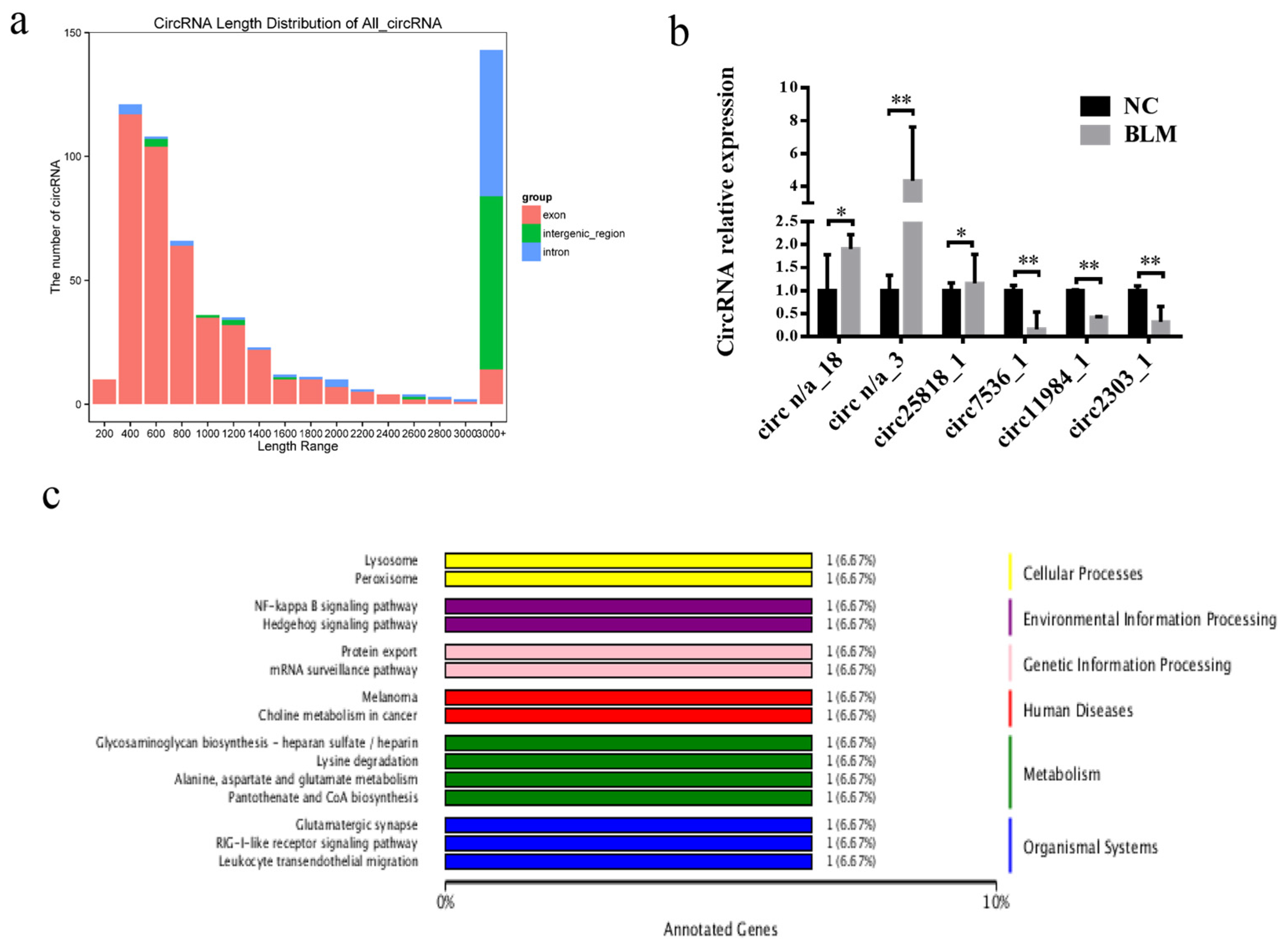
3.5. Differentially Expressed circRNA Identification
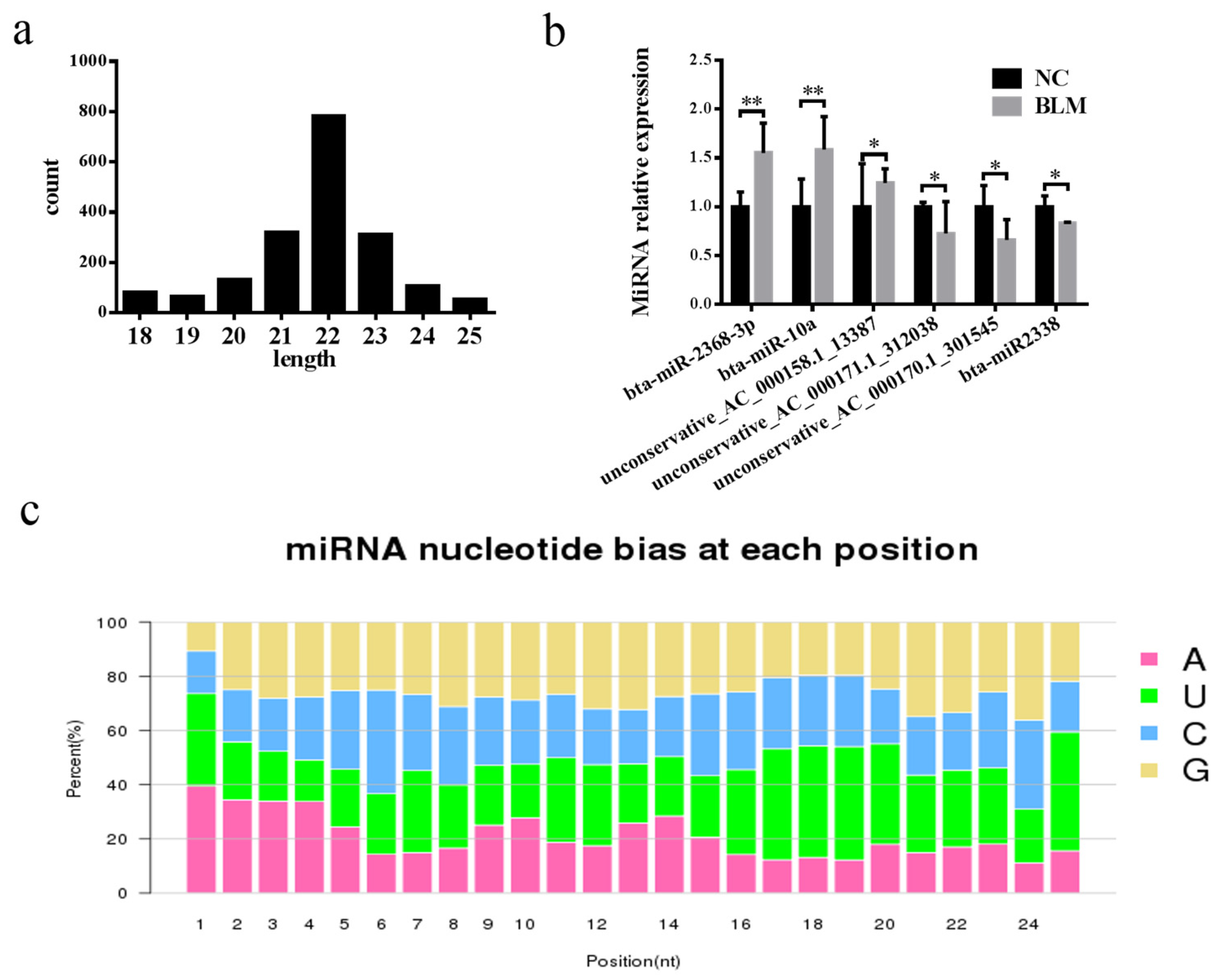
3.6. Differentially Expressed miRNA Identification
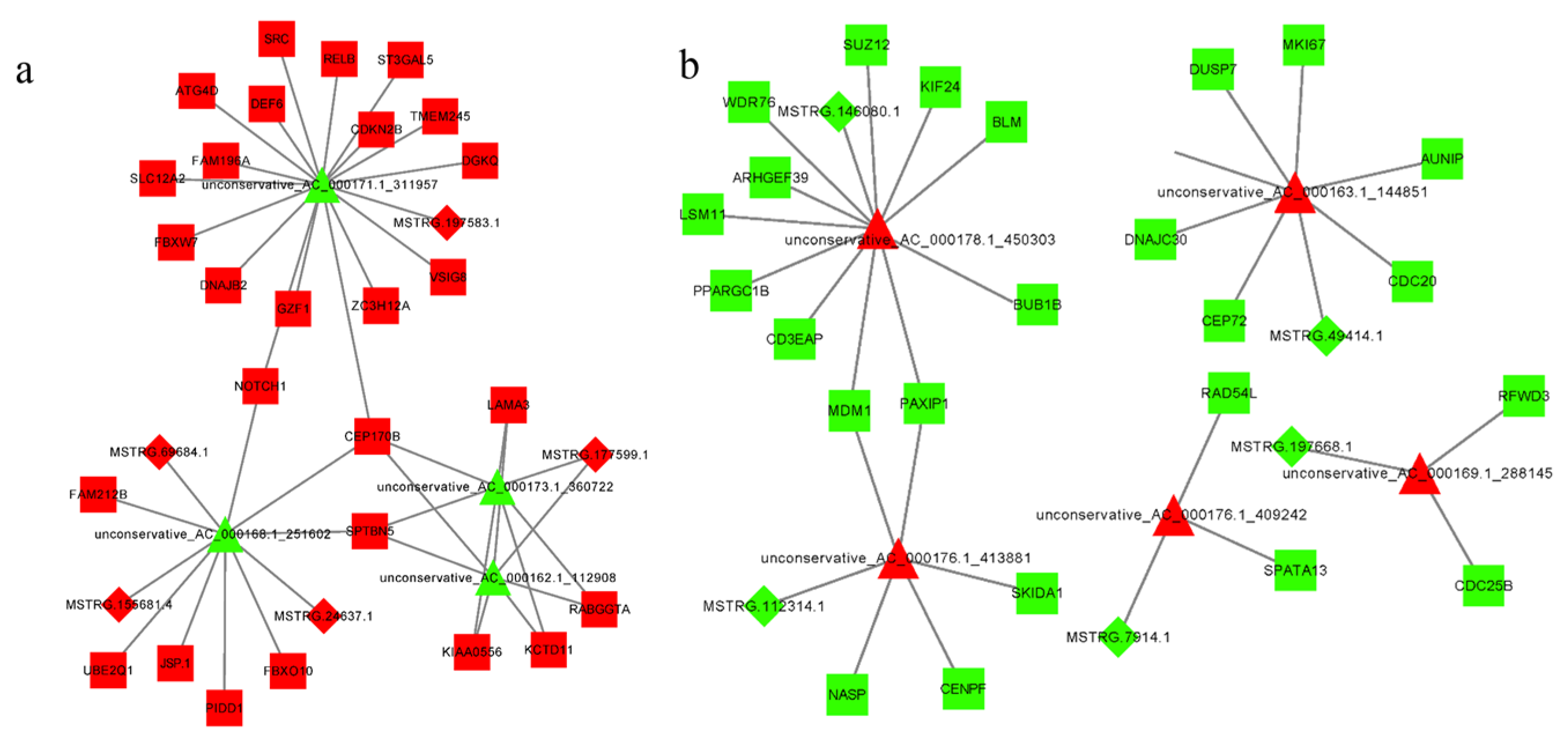
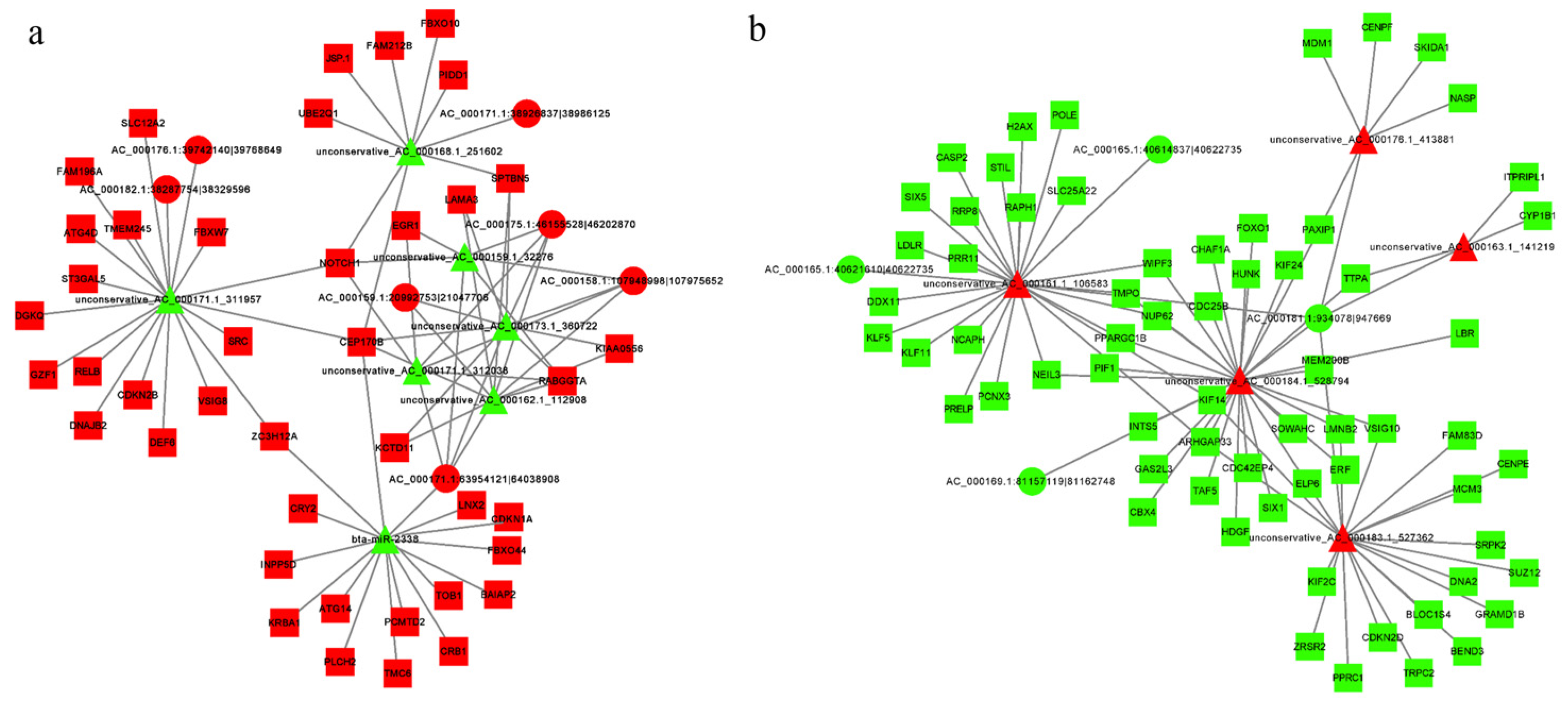
3.7. Construction of the lncRNA–miRNA–mRNA Regulatory Network
3.8. Construction of the circRNA–miRNA–mRNA Regulatory Network
3.9. Construction of the LncRNA/CircRNA–miRNA–mRNA Regulatory Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buccione, R.; Vanderhyden, B.C.; Caron, P.J.; Eppig, J.J. FSH-induced expansion of the mouse cumulus oophorus in vitro is dependent upon a specific factor(s) secreted by the oocyte. Dev. Biol. 1990, 138, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.X. Interaction and signal transduction between oocyte and samatic cells in the ovary. Front. Biosci. 2007, 12, 2782–2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.Q.; Sugiura, K.; Eppig, J.J. Mouse oocyte control of granulosa cell development and function: Paracrine regulation of cumulus cell metabolism. Semin. Reprod. Med. 2009, 27, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Neal, J.A.; Meek, K. Choosing the right path: Does DNA-PK help make the decision? Mutat. Res. 2011, 711, 73–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, A.J.; Chen, B.P.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Tu, W.Z.; Li, B.; Huang, B.; Wang, Y.; Liu, X.D.; Guan, H.; Zhang, S.M.; Tang, Y.; Rang, W.Q.; Zhou, P.K. gammaH2AX foci formation in the absence of DNA damage: Mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS Lett. 2013, 587, 3437–3443. [Google Scholar] [CrossRef] [Green Version]
- Pandita, T.K.; Richardson, C. Chromatin remodeling finds its place in the DNA double-strand break response. Nucleic Acids Res. 2009, 37, 1363–1377. [Google Scholar] [CrossRef]
- van Gent, D.C.; Hoeijmakers, J.H.; Kanaar, R. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet. 2001, 2, 196–206. [Google Scholar] [CrossRef]
- Bohgaki, T.; Bohgaki, M.; Hakem, R. DNA double-strand break signaling and human disorders. Genome Integr. 2010, 1, 15. [Google Scholar] [CrossRef] [Green Version]
- Raman, R.S.; Chan, P.J.; Corselli, J.U.; Patton, W.C.; Jacobson, J.D.; Chan, S.R.; King, A. Comet assay of cumulus cell DNA status and the relationship to oocyte fertilization via intracytoplasmic sperm injection. Hum. Reprod. 2001, 16, 831–835. [Google Scholar] [CrossRef]
- Tuppi, M.; Kehrloesser, S.; Coutandin, D.W.; Rossi, V.; Luh, L.M.; Strubel, A.; Hotte, K.; Hoffmeister, M.; Schafer, B.; De Oliveira, T.; et al. Oocyte DNA damage quality control requires consecutive interplay of CHK2 and CK1 to activate p63. Nat. Struct. Mol. Biol. 2018, 25, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Horta, F.; Catt, S.; Ramachandran, P.; Vollenhoven, B.; Temple-Smith, P. Female ageing affects the DNA repair capacity of oocytes in IVF using a controlled model of sperm DNA damage in mice. Hum. Reprod. 2020, 35, 529–544. [Google Scholar] [CrossRef] [PubMed]
- Marangos, P.; Carroll, J. Oocytes progress beyond prophase in the presence of DNA damage. Curr. Biol. 2012, 22, 989–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Luo, Y.; Zhao, M.H.; Lin, Z.; Kwon, J.; Cui, X.S.; Kim, N.H. DNA double-strand breaks disrupted the spindle assembly in porcine oocytes. Mol. Reprod. Dev. 2016, 83, 132–143. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, G.L.; Ma, J.Y.; Qi, S.T.; Wang, Z.B.; Wang, Z.W.; Luo, Y.B.; Jiang, Z.Z.; Schatten, H.; Sun, Q.Y. Effects of DNA damage and short-term spindle disruption on oocyte meiotic maturation. Histochem. Cell Biol. 2014, 142, 185–194. [Google Scholar] [CrossRef]
- Wang, L.; Xu, X.; Teng, M.; Zhao, G.; Lei, A. Coping with DNA Double-Strand Breaks via ATM Signaling Pathway in Bovine Oocytes. Int. J. Mol. Sci. 2020, 21, 8892. [Google Scholar] [CrossRef]
- Yamamoto, T.; Iwata, H.; Goto, H.; Shiratuki, S.; Tanaka, H.; Monji, Y.; Kuwayama, T. Effect of maternal age on the developmental competence and progression of nuclear maturation in bovine oocytes. Mol. Reprod. Dev. 2010, 77, 595–604. [Google Scholar] [CrossRef]
- Sun, M.H.; Zheng, J.; Xie, F.Y.; Shen, W.; Yin, S.; Ma, J.Y. Cumulus Cells Block Oocyte Meiotic Resumption via Gap Junctions in Cumulus Oocyte Complexes Subjected to DNA Double-Strand Breaks. PLoS ONE 2015, 10, e0143223. [Google Scholar] [CrossRef]
- Sirini, M.A.; Anchordoquy, J.M.; Anchordoquy, J.P.; Pascua, A.M.; Nikoloff, N.; Carranza, A.; Relling, A.E.; Furnus, C.C. The presence of acylated ghrelin during in vitro maturation of bovine oocytes induces cumulus cell DNA damage and apoptosis, and impairs early embryo development. Zygote 2017, 25, 601–611. [Google Scholar] [CrossRef]
- Takahashi, M.; Keicho, K.; Takahashi, H.; Ogawa, H.; Schultz, R.M.; Okano, A. Effect of oxidative stress on development and DNA damage in in-vitro cultured bovine embryos by comet assay. Theriogenology 2000, 54, 137–145. [Google Scholar] [CrossRef]
- Sun, M.H.; Yang, M.; Xie, F.Y.; Wang, W.; Zhang, L.; Shen, W.; Yin, S.; Ma, J.Y. DNA Double-Strand Breaks Induce the Nuclear Actin Filaments Formation in Cumulus-Enclosed Oocytes but Not in Denuded Oocytes. PLoS ONE 2017, 12, e0170308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turan, V.; Oktay, K. BRCA-related ATM-mediated DNA double-strand break repair and ovarian aging. Hum. Reprod. Update 2020, 26, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Maidarti, M.; Anderson, R.A.; Telfer, E.E. Crosstalk between PTEN/PI3K/Akt Signalling and DNA Damage in the Oocyte: Implications for Primordial Follicle Activation, Oocyte Quality and Ageing. Cells 2020, 9, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, V.D.; Bloom, J.C.; Schimenti, J.C. Oocyte Elimination through DNA Damage Signaling from CHK1/CHK2 to p53 and p63. Genetics 2020, 215, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [Green Version]
- Guil, S.; Esteller, M. RNA-RNA interactions in gene regulation: The coding and noncoding players. Trends Biochem. Sci. 2015, 40, 248–256. [Google Scholar] [CrossRef]
- Lian, Y.; Yan, C.; Xu, H.; Yang, J.; Yu, Y.; Zhou, J.; Shi, Y.; Ren, J.; Ji, G.; Wang, K. A Novel lncRNA, LINC00460, Affects Cell Proliferation and Apoptosis by Regulating KLF2 and CUL4A Expression in Colorectal Cancer. Mol. Nucleic Acids 2018, 12, 684–697. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Guo, B.; Li, Q.; Peng, J.; Yang, Z.; Wang, A.; Li, D.; Hou, Z.; Lv, K.; Kan, G.; et al. miR-214 targets ATF4 to inhibit bone formation. Nat. Med. 2013, 19, 93–100. [Google Scholar] [CrossRef]
- Zhang, K.; Han, X.; Zhang, Z.; Zheng, L.; Hu, Z.; Yao, Q.; Cui, H.; Shu, G.; Si, M.; Li, C.; et al. The liver-enriched lnc-LFAR1 promotes liver fibrosis by activating TGFbeta and Notch pathways. Nat. Commun. 2017, 8, 144. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Chen, T.; Yao, Q.; Zheng, L.; Zhang, Z.; Wang, J.; Hu, Z.; Cui, H.; Han, Y.; Han, X.; et al. The circular RNA ciRS-7 promotes APP and BACE1 degradation in an NF-kappaB-dependent manner. FEBS J. 2017, 284, 1096–1109. [Google Scholar] [CrossRef]
- Gilchrist, G.C.; Tscherner, A.; Nalpathamkalam, T.; Merico, D.; LaMarre, J. MicroRNA Expression during Bovine Oocyte Maturation and Fertilization. Int. J. Mol. Sci. 2016, 17, 396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Niu, Z.; Li, Q.; Pang, R.T.; Chiu, P.C.; Yeung, W.S. MicroRNA and Embryo Implantation. Am. J. Reprod. Immunol. 2016, 75, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Dehghan, Z.; Mohammadi-Yeganeh, S.; Salehi, M. MiRNA-155 regulates cumulus cells function, oocyte maturation, and blastocyst formation. Biol. Reprod. 2020, 103, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, H.; Sheng, Y.; Wang, Z. MicroRNA-224 delays oocyte maturation through targeting Ptx3 in cumulus cells. Mech. Dev. 2017, 143, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Iyyappan, R.; Aleshkina, D.; Zhu, L.; Jiang, Z.; Kinterova, V.; Susor, A. Oocyte specific lncRNA variant Rose influences oocyte and embryo development. Noncoding RNA Res. 2021, 6, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Gao, D.; Xu, T.; Zhang, L.; Tong, X.; Zhang, D.; Wang, Y.; Ning, W.; Qi, X.; Ma, Y.; et al. Circular RNA profiling in the oocyte and cumulus cells reveals that circARMC4 is essential for porcine oocyte maturation. Aging 2019, 11, 8015–8034. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Wang, Q.; Liu, R.; Chen, Z.; Zhang, X.; Zhou, P.; Wang, Z. LncRNA lnc-RI regulates homologous recombination repair of DNA double-strand breaks by stabilizing RAD51 mRNA as a competitive endogenous RNA. Nucleic Acids Res. 2018, 46, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Deng, T.; Ge, S.; Liu, Y.; Bai, M.; Zhu, K.; Fan, Q.; Li, J.; Ning, T.; Tian, F.; et al. Exosome circRNA secreted from adipocytes promotes the growth of hepatocellular carcinoma by targeting deubiquitination-related USP7. Oncogene 2018, 38, 2844–2859. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Stubbe, J. Bleomycins: Towards better therapeutics. Nat. Rev. Cancer 2005, 5, 102–112. [Google Scholar] [CrossRef]
- Cloos, J.; Temmink, O.; Ceelen, M.; Snel, M.H.; Leemans, C.R.; Braakhuis, B.J. Involvement of cell cycle control in bleomycin-induced mutagen sensitivity. Environ. Mol. Mutagen. 2002, 40, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, T.; Lei, Y.; Li, X.; Jiang, J.; Lan, J.; Liu, Y.; Chen, H.; Gao, W.; Xie, N.; et al. Identification of ANXA2 (annexin A2) as a specific bleomycin target to induce pulmonary fibrosis by impeding TFEB-mediated autophagic flux. Autophagy 2018, 14, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Groselj, A.; Krzan, M.; Kosjek, T.; Bosnjak, M.; Sersa, G.; Cemazar, M. Bleomycin pharmacokinetics of bolus bleomycin dose in elderly cancer patients treated with electrochemotherapy. Cancer Chemother. Pharmacol. 2016, 77, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, C.; Jiang, H.; Gao, Y.; Xu, M.; Wang, J.; Liu, S.; Fu, Y.; Sun, X.; Xu, J.; et al. Regulatory Role of miRNA-375 in Expression of BMP15/GDF9 Receptors and its Effect on Proliferation and Apoptosis of Bovine Cumulus Cells. Cell. Physiol. Biochem. 2017, 41, 439–450. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Wang, C.; Sun, H.; Wang, J.; Liang, Y.; Wang, Y.; Wong, G. The bioinformatics toolbox for circRNA discovery and analysis. Brief. Bioinform. 2021, 22, 1706–1728. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, L.; Wang, J.; Gu, P.; Chen, M. MTide: An integrated tool for the identification of miRNA-target interaction in plants. Bioinformatics 2015, 31, 290–291. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Xue, J.; Sun, B.; Zou, Z.; Chen, C.; Liu, Q.; Zhang, A. miR-145 via targeting ERCC2 is involved in arsenite-induced DNA damage in human hepatic cells. Toxicol. Lett. 2018, 295, 220–228. [Google Scholar] [CrossRef]
- Mocavini, I.; Pippa, S.; Licursi, V.; Paci, P.; Trisciuoglio, D.; Mannironi, C.; Presutti, C.; Negri, R. JARID1B expression and its function in DNA damage repair are tightly regulated by miRNAs in breast cancer. Cancer Sci. 2018, 110, 1232–1243. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Zhang, L.; Xu, J.; Zhu, C.; Zhao, H.; Zhu, Y.; Lv, G. AntogomiR-451 protects human gastric epithelial cells from ethanol via activating AMPK signaling. Biochem. Biophys. Res. Commun. 2018, 497, 339–346. [Google Scholar] [CrossRef]
- Banath, J.P.; Olive, P.L. Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double-strand breaks. Cancer Res. 2003, 63, 4347–4350. [Google Scholar]
- Stucki, M.; Jackson, S.P. gammaH2AX and MDC1: Anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair 2006, 5, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Oosthuysen, W.F.; Mueller, T.; Dittrich, M.T.; Schubert-Unkmeir, A. Neisseria meningitidis causes cell cycle arrest of human brain microvascular endothelial cells at S phase via p21 and cyclin G2. Cell. Microbiol. 2016, 18, 46–65. [Google Scholar] [CrossRef] [PubMed]
- Mahaddalkar, T.; Mehta, S.; Cheriyamundath, S.; Muthurajan, H.; Lopus, M. Tryptone-stabilized gold nanoparticles target tubulin and inhibit cell viability by inducing an unusual form of cell cycle arrest. Exp. Cell Res. 2017, 360, 163–170. [Google Scholar] [CrossRef]
- Stillman, B. Cell cycle control of DNA replication. Science 1996, 274, 1659–1664. [Google Scholar] [CrossRef]
- Shaltiel, I.A.; Krenning, L.; Bruinsma, W.; Medema, R.H. The same, only different—DNA damage checkpoints and their reversal throughout the cell cycle. J. Cell Sci. 2015, 128, 607–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Miao, Z.H.; Zhu, H.; Cai, Y.J.; Lu, W.; Ding, J. Chk1 and Chk2 are differentially involved in homologous recombination repair and cell cycle arrest in response to DNA double-strand breaks induced by camptothecins. Mol. Cancer Ther. 2008, 7, 1440–1449. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Xu, Y.; Roy, K.; Price, B.D. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol. Cell. Biol. 2007, 27, 8502–8509. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Hou, Y.; Zhang, H.; Tu, G.; Yang, L.; Sun, Y.; Lang, L.; Tang, X.; Du, Y.E.; Zhou, M.; et al. Oxidized ATM promotes abnormal proliferation of breast CAFs through maintaining intracellular redox homeostasis and activating the PI3K-AKT, MEK-ERK, and Wnt-beta-catenin signaling pathways. Cell Cycle 2015, 14, 1908–1924. [Google Scholar] [CrossRef]
- Schotz, U.; Balzer, V.; Brandt, F.W.; Ziemann, F.; Subtil, F.S.B.; Rieckmann, T.; Kocher, S.; Engenhart-Cabillic, R.; Dikomey, E.; Wittig, A.; et al. Dual PI3K/mTOR Inhibitor NVP-BEZ235 Enhances Radiosensitivity of Head and Neck Squamous Cell Carcinoma (HNSCC) Cell Lines Due to Suppressed Double-Strand Break (DSB) Repair by Non-Homologous End Joining. Cancers 2020, 12, 467. [Google Scholar] [CrossRef] [Green Version]
- Beermann, J.; Piccoli, M.T.; Viereck, J.; Thum, T. Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, F.; Albertini, M.C.; Orciani, M.; Ceka, A.; Cricca, M.; Procopio, A.D.; Bonafe, M. DNA damage response (DDR) and senescence: Shuttled inflamma-miRNAs on the stage of inflamm-aging. Oncotarget 2015, 6, 35509–35521. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.M.; Garcia, J.T.; Hung, T.; Flynn, R.A.; Shen, Y.; Qu, K.; Payumo, A.Y.; Peres-da-Silva, A.; Broz, D.K.; Baum, R.; et al. An inducible long noncoding RNA amplifies DNA damage signaling. Nat. Genet. 2016, 48, 1370–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shihabudeen Haider Ali, M.S.; Cheng, X.; Moran, M.; Haemmig, S.; Naldrett, M.J.; Alvarez, S.; Feinberg, M.W.; Sun, X. LncRNA Meg3 protects endothelial function by regulating the DNA damage response. Nucleic Acids Res. 2018, 47, 1505–1522. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Morselli, E.; Vitale, I.; Kepp, O.; Senovilla, L.; Criollo, A.; Servant, N.; Paccard, C.; Hupe, P.; Robert, T.; et al. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res. 2010, 70, 1793–1803. [Google Scholar] [CrossRef] [Green Version]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Lee, J.H.; Ha, M.; Nam, J.W.; Kim, V.N. miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat. Struct. Mol. Biol. 2009, 16, 23–29. [Google Scholar] [CrossRef]
- Le, M.T.; Teh, C.; Shyh-Chang, N.; Xie, H.; Zhou, B.; Korzh, V.; Lodish, H.F.; Lim, B. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009, 23, 862–876. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.Y.; Li, Y.J.; Wang, P.Y.; Jiao, F.; Zhang, S.; Zhang, W.J. miRNA-regulated expression of oncogenes and tumor suppressor genes in the cisplatin-inhibited growth of K562 cells. Oncol. Rep. 2010, 23, 1693–1700. [Google Scholar] [CrossRef]
- Zhang, Q.; Mady, A.S.A.; Ma, Y.; Ryan, C.; Lawrence, T.S.; Nikolovska-Coleska, Z.; Sun, Y.; Morgan, M.A. The WD40 domain of FBXW7 is a poly(ADP-ribose)-binding domain that mediates the early DNA damage response. Nucleic Acids Res. 2019, 47, 4039–4053. [Google Scholar] [CrossRef]
- Zhang, P.; Cao, L.; Fan, P.; Mei, Y.; Wu, M. LncRNA-MIF, a c-Myc-activated long non-coding RNA, suppresses glycolysis by promoting Fbxw7-mediated c-Myc degradation. EMBO Rep. 2016, 17, 1204–1220. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ma, J.; Xu, F.; Li, L. TINCR suppresses proliferation and invasion through regulating miR-544a/FBXW7 axis in lung cancer. Biomed. Pharmacother. 2018, 99, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Tinel, A.; Lippens, S.; Tschopp, J. PIDD mediates NF-kappaB activation in response to DNA damage. Cell 2005, 123, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- De Gregoriis, G.; Ramos, J.A.; Fernandes, P.V.; Vignal, G.M.; Brianese, R.C.; Carraro, D.M.; Monteiro, A.N.; Struchiner, C.J.; Suarez-Kurtz, G.; Vianna-Jorge, R.; et al. DNA repair genes PAXIP1 and TP53BP1 expression is associated with breast cancer prognosis. Cancer Biol. Ther. 2017, 18, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Inano, S.; Sato, K.; Katsuki, Y.; Kobayashi, W.; Tanaka, H.; Nakajima, K.; Nakada, S.; Miyoshi, H.; Knies, K.; Takaori-Kondo, A.; et al. RFWD3-Mediated Ubiquitination Promotes Timely Removal of Both RPA and RAD51 from DNA Damage Sites to Facilitate Homologous Recombination. Mol. Cell. 2017, 66, 622–634.e8. [Google Scholar] [CrossRef] [Green Version]
- Ricciuti, B.; Recondo, G.; Spurr, L.F.; Li, Y.Y.; Lamberti, G.; Venkatraman, D.; Umeton, R.; Cherniack, A.D.; Nishino, M.; Sholl, L.M.; et al. Impact of DNA Damage Response and Repair (DDR) Gene Mutations on Efficacy of PD-(L)1 Immune Checkpoint Inhibition in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 4135–4142. [Google Scholar] [CrossRef] [Green Version]
- Abbotts, R.; Thompson, N.; Madhusudan, S. DNA repair in cancer: Emerging targets for personalized therapy. Cancer Manag. Res. 2014, 6, 77–92. [Google Scholar] [CrossRef] [Green Version]
- Carlsen, L.; El-Deiry, W.S. Anti-cancer immune responses to DNA damage response inhibitors: Molecular mechanisms and progress toward clinical translation. Front. Oncol. 2022, 12, 998388. [Google Scholar] [CrossRef] [PubMed]
- Ruth, K.S.; Day, F.R.; Hussain, J.; Martinez-Marchal, A.; Aiken, C.E.; Azad, A.; Thompson, D.J.; Knoblochova, L.; Abe, H.; Tarry-Adkins, J.L.; et al. Genetic insights into biological mechanisms governing human ovarian ageing. Nature 2021, 596, 393–397. [Google Scholar] [CrossRef]
- Shaw, A.; Gullerova, M. Home and Away: The Role of Non-Coding RNA in Intracellular and Intercellular DNA Damage Response. Genes 2021, 12, 1475. [Google Scholar] [CrossRef] [PubMed]
- Cerna, K.; Oppelt, J.; Chochola, V.; Musilova, K.; Seda, V.; Pavlasova, G.; Radova, L.; Arigoni, M.; Calogero, R.A.; Benes, V.; et al. MicroRNA miR-34a downregulates FOXP1 during DNA damage response to limit BCR signalling in chronic lymphocytic leukaemia B cells. Leukemia 2019, 33, 403–414. [Google Scholar] [CrossRef]
- Michelini, F.; Pitchiaya, S.; Vitelli, V.; Sharma, S.; Gioia, U.; Pessina, F.; Cabrini, M.; Wang, Y.; Capozzo, I.; Iannelli, F.; et al. Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat. Cell Biol. 2017, 19, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Altmeyer, M. A lncRNA to repair DNA. EMBO Rep. 2015, 16, 1413–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, M.; Zhang, N.; Yao, S.; Zhou, H.; Chen, X.; Jia, Y.; Zhang, H.; Li, X.; Jiang, Y. DNMT3A-mediated high expression of circ_0057504 promotes benzo[a]pyrene-induced DNA damage via the NONO-SFPQ complex in human bronchial epithelial cells. Environ. Int. 2022, 170, 107627. [Google Scholar] [CrossRef] [PubMed]
- Li, P.P.; Li, R.G.; Huang, Y.Q.; Lu, J.P.; Zhang, W.J.; Wang, Z.Y. LncRNA OTUD6B-AS1 promotes paclitaxel resistance in triple negative breast cancer by regulation of miR-26a-5p/MTDH pathway-mediated autophagy and genomic instability. Aging 2021, 13, 24171–24191. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.-B.; Zhang, J.-B.; Yan, X.-M.; Xie, P.-G.; Fu, Y.; Fu, X.-H.; Sun, X.-L.; Han, D.-X.; Li, S.-P.; Zheng, Y.; et al. DNA Double-Strand Break-Related Competitive Endogenous RNA Network of Noncoding RNA in Bovine Cumulus Cells. Genes 2023, 14, 290. https://doi.org/10.3390/genes14020290
Liu J-B, Zhang J-B, Yan X-M, Xie P-G, Fu Y, Fu X-H, Sun X-L, Han D-X, Li S-P, Zheng Y, et al. DNA Double-Strand Break-Related Competitive Endogenous RNA Network of Noncoding RNA in Bovine Cumulus Cells. Genes. 2023; 14(2):290. https://doi.org/10.3390/genes14020290
Chicago/Turabian StyleLiu, Jian-Bo, Jia-Bao Zhang, Xiang-Min Yan, Peng-Gui Xie, Yao Fu, Xu-Huang Fu, Xu-Lei Sun, Dong-Xu Han, Sheng-Peng Li, Yi Zheng, and et al. 2023. "DNA Double-Strand Break-Related Competitive Endogenous RNA Network of Noncoding RNA in Bovine Cumulus Cells" Genes 14, no. 2: 290. https://doi.org/10.3390/genes14020290