
Treatment Dilemma in Children with Late-Onset Pompe Disease
 , , , and
, , , and 
Abstract
:1. Introduction
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chan, J.; Desai, A.K.; Kazi, Z.B.; Corey, K.; Austin, S.; Hobson-Webb, L.D.; Case, L.E.; Jones, H.N.; Kishnani, P.S. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol. Genet. Metab. 2017, 120, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, C.; Papadimas, G.K.; Michelakakis, H.; Kararizou, E.; Manta, P. Highlighting intrafamilial clinical heterogeneity in late-onset Pompe disease. Mol. Genet. Metab. Rep. 2014, 1, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Ausems, M.G.E.M.; Ten Berg, K.; Beemer, F.A.; Wokke, J.H.J. Phenotypic expression of late-onset glycogen storage disease type II: Identification of asymptomatic adults through family studies and review of reported families. Neuromuscul. Disord. 2000, 10, 467–471. [Google Scholar] [CrossRef]
- Martínez, J.R.B.; Martínez, A.C. Long-term enzyme-replacement therapy (ERT) with alglucosidase alfa: Evolution of two siblings with juvenile late-onset Pompe disease. J. Neurol. Sci. 2015, 358, 459–460. [Google Scholar] [CrossRef]
- van der Ploeg, A.T.; Clemens, P.R.; Corzo, D.; Escolar, D.M.; Florence, J.; Groeneveld, G.J.; Herson, S.; Kishnani, P.S.; Laforet, P.; Lake, S.L.; et al. A Randomized Study of Alglucosidase Alfa in Late-Onset Pompe’s Disease. N. Engl. J. Med. 2010, 362, 1396–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishnani, P.S.; Corzo, D.; Nicolino, M.; Byrne, B.; Mandel, H.; Hwu, W.-L.; Leslie, N.; Levine, J.; Spencer, C.; McDonald, M.; et al. Recombinant human acid α-glucosidase: Major clinical benefits in infantile-onset Pompe disease. Neurology 2007, 68, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Dornelles, A.D.; Junges, A.P.P.; Pereira, T.V.; Krug, B.C.; Gonçalves, C.B.T.; Llerena, J.C.; Kishnani, P.S.; de Oliveira, H.A.; Schwartz, I.V.D. A systematic review and meta-analysis of enzyme replacement therapy in late-onset pompe disease. J. Clin. Med. 2021, 10, 4828. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Goldenberg, P.C.; DeArmey, S.L.; Heller, J.; Benjamin, D.; Young, S.; Bali, D.; Smith, S.A.; Li, J.S.; Mandel, H.; et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol. Genet. Metab. 2010, 99, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desnick, R.J.; Schuchman, E.H. Enzyme Replacement Therapy for Lysosomal Diseases: Lessons from 20 Years of Experience and Remaining Challenges. Hum. Genet. 2012, 13, 307–335. [Google Scholar] [CrossRef] [Green Version]
- Llerena, J.C., Jr.; Nascimento, O.J.; Oliveira, A.S.B.; Dourado, M.E.T., Jr.; Marrone, C.D.; Siqueira, H.H.; Sobreira, C.F.R.; Dias-Tosta, E.; Werneck, L.C. Guidelines for the diagnosis, treatment and clinical monitoring of patients with juvenile and adult pompe disease. Arq. Neuropsiquiatr. 2016, 74, 166–176. [Google Scholar] [CrossRef] [Green Version]
- de Vries, J.M.; Kuperus, E.; Hoogeveen-Westerveld, M.; Kroos, M.A.; Wens, S.C.; Stok, M.; van der Beek, N.A.; Kruijshaar, M.E.; Rizopoulos, D.; van Doorn, P.A.; et al. Pompe disease in adulthood: Effects of antibody formation on enzyme replacement therapy. Genet. Med. 2017, 19, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Manera, J.; Kishnani, P.S.; Kushlaf, H.; Ladha, S.; Mozaffar, T.; Straub, V.; Toscano, A.; van der Ploeg, A.T.; Berger, K.I.; Clemens, P.R.; et al. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late-onset Pompe disease (COMET): A phase 3, randomised, multicentre trial. Lancet Neurol. 2021, 20, 1012–1026. [Google Scholar] [CrossRef] [PubMed]
- Kishnani, P.S.; Sun, B.; Koeberl, D.D. Gene therapy for glycogen storage diseases. Hum. Mol. Genet. 2019, 28, R31–R41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoser, B.; Roberts, M.; Byrne, B.J.; Sitaraman, S.; Jiang, H.; Laforêt, P.; Toscano, A.; Castelli, J.; Díaz-Manera, J.; Goldman, M.; et al. Safety and efficacy of cipaglucosidase alfa plus miglustat versus alglucosidase alfa plus placebo in late-onset Pompe disease (PROPEL): An international, randomised, double-blind, parallel-group, phase 3 trial. Lancet Neurol. 2021, 20, 1027–1037. [Google Scholar] [CrossRef]
- Niu, D.-M.; Lin, H.-Y.; Wu, T.J.-T.; Hsu, J.-H.; Yu, H.-C.; Lin, S.-P.; Chuang, C.-K.; Huang, C.-H. Novel human pathological mutations. Gene symbol: GAA. Disease: Glycogen storage disease 2. Hum. Genet. 2010, 127, 465. [Google Scholar]
- Niño, M.Y.; In’t Groen, S.L.; Bergsma, A.J.; van der Beek, N.A.; Kroos, M.; Hoogeveen-Westerveld, M.; Van Der Ploeg, A.T.; Pijnappel, W.P. Extension of the Pompe mutation database by linking disease-associated variants to clinical severity. Hum. Mutat. 2019, 40, 1954–1967. [Google Scholar] [CrossRef] [Green Version]
- Romero, M.B.; Cortes, E.B.; Lorite, J.B.; Rivas, E.G.; Sendra, I.I.; Jiménez, L.M.; Martos, M.L.; Arregui, A.L.D.M.; Fernández, J.P.; Pascual, S.I.P.; et al. Guía clínica de la enfermedad de Pompe de inicio tardío. Rev. Neurol. 2012, 54, 497. [Google Scholar] [CrossRef] [Green Version]
- Schüller, A.; Kornblum, C.; Deschauer, M.; Vorgerd, M.; Schrank, B.; Mengel, E.; Lukacs, Z.; Gläser, D.; Young, P.; Plöckinger, U.; et al. Diagnose und Therapie des Late-onset-Morbus-Pompe. Nervenarzt 2013, 84, 1467–1472. [Google Scholar] [CrossRef]
- Hundsberger, T.; Rohrbach, M.; Kern, L.; Rösler, K.M. Swiss national guideline for reimbursement of enzyme replacement therapy in late-onset Pompe disease. J. Neurol. 2013, 260, 2279–2285. [Google Scholar] [CrossRef]
- Yang, C.-F.; Yang, C.C.; Liao, H.-C.; Huang, L.-Y.; Chiang, C.-C.; Ho, H.-C.; Lai, C.-J.; Chu, T.-H.; Yang, T.-F.; Hsu, T.; et al. Very Early Treatment for Infantile-Onset Pompe Disease Contributes to Better Outcomes. J. Pediatr. 2016, 169, 174–180.e1. [Google Scholar] [CrossRef]
- Laloui, K.; Wary, C.; Carlier, R.-Y.; Hogrel, J.-Y.; Caillaud, C.; Laforet, P. Making diagnosis of Pompe disease at a presymptomatic stage: To treat or not to treat? Neurology 2011, 77, 594–595. [Google Scholar] [CrossRef] [PubMed]
- Echaniz-Laguna, A.; Carlier, R.-Y.; Laloui, K.; Carlier, P.; Salort-Campana, E.; Pouget, J.; Laforet, P. Should patients with asymptomatic pompe disease be treated? A nationwide study in France. Muscle Nerve 2015, 51, 884–889. [Google Scholar] [CrossRef] [PubMed]
- Ashe, K.M.; Taylor, K.M.; Chu, Q.; Meyers, E.; Ellis, A.; Jingozyan, V.; Klinger, K.; Finn, P.F.; Cooper, C.G.; Chuang, W.-L.; et al. Inhibition of glycogen biosynthesis via mTORC1 suppression as an adjunct therapy for Pompe disease. Mol. Genet. Metab. 2010, 100, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Li, L.; Shirihai, O.S.; Trudeau, K.M.; Puertollano, R.; Raben, N. Modulation of mTOR signaling as a strategy for the treatment of Pompe disease. EMBO Mol. Med. 2017, 9, 353–370. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.-W.; Xu, X.-B.; Ji, K.-Q.; Zhang, N.; Sun, Y.; Zhao, D.-D.; Zhao, Y.-Y.; Yan, C.-Z. Activated mTOR signaling pathway in myofibers with inherited metabolic defect might be an evidence for mTOR inhibition therapies. Chin. Med. J. 2019, 132, 805–810. [Google Scholar] [CrossRef]
- Kronn, D.F.; Day-Salvatore, D.; Hwu, W.-L.; Jones, S.A.; Nakamura, K.; Okuyama, T.; Swoboda, K.J.; Kishnani, P.S. Management of confirmed newborn-screened patients with pompe disease across the disease spectrum. Pediatrics 2017, 140, S24–S45. [Google Scholar] [CrossRef] [Green Version]
- van der Ploeg, A.T.; Kruijshaar, M.E.; Toscano, A.; Laforêt, P.; Angelini, C.; Lachmann, R.H.; Pascual, S.I.P.; Roberts, M.; Rösler, K.; Stulnig, T.; et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: A 10-year experience. Eur. J. Neurol. 2017, 24, 768-e31. [Google Scholar] [CrossRef]
- Al Jasmi, F.; Al Jumah, M.; Alqarni, F.; Al-Sanna’A, N.; Al-Sharif, F.; Bohlega, S.; Cupler, E.J.; Fathalla, W.; Hamdan, M.A. Diagnosis and treatment of late-onset Pompe disease in the Middle East and North Africa region: Consensus recommendations from an expert group. BMC Neurol. 2015, 15, 205. [Google Scholar] [CrossRef]
- Pichiecchio, A.; Uggetti, C.; Ravaglia, S.; Egitto, M.G.; Rossi, M.; Sandrini, G.; Danesino, C. Muscle MRI in adult-onset acid maltase deficiency. Neuromuscul. Disord. 2004, 14, 51–55. [Google Scholar] [CrossRef]
- Khan, A.; Boggs, T.; Rt, M.B.; Austin, S.; Bs, M.S.; Case, L.; Kishnani, P.S. Whole-body magnetic resonance imaging in late-onset Pompe disease: Clinical utility and correlation with functional measures. J. Inherit. Metab. Dis. 2020, 43, 549–557. [Google Scholar] [CrossRef]
- Díaz-Manera, J.; Walter, G.; Straub, V. Skeletal muscle magnetic resonance imaging in Pompe disease. Muscle Nerve 2021, 63, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Figueroa-Bonaparte, S.; Segovia, S.; Llauger, J.; Belmonte, I.; Pedrosa, I.; Alejaldre, A.; Mayos, M.; Suárez-Cuartín, G.; Gallardo, E.; Illa, I.; et al. Muscle MRI findings in childhood/adult onset pompe disease correlate with muscle function. PLoS ONE 2016, 11, e0163493. [Google Scholar] [CrossRef] [PubMed]
- Pichiecchio, A.; Poloni, G.; Ravaglia, S.; Ponzio, M.; Germani, G.; Maranzana, D.; Costa, A.; Repetto, A.; Tavazzi, E.; Danesino, C.; et al. Enzyme replacement therapy in adult-onset glycogenosis II: Is quantitative muscle MRI helpful? Muscle Nerve 2009, 40, 122–125. [Google Scholar] [CrossRef] [PubMed]
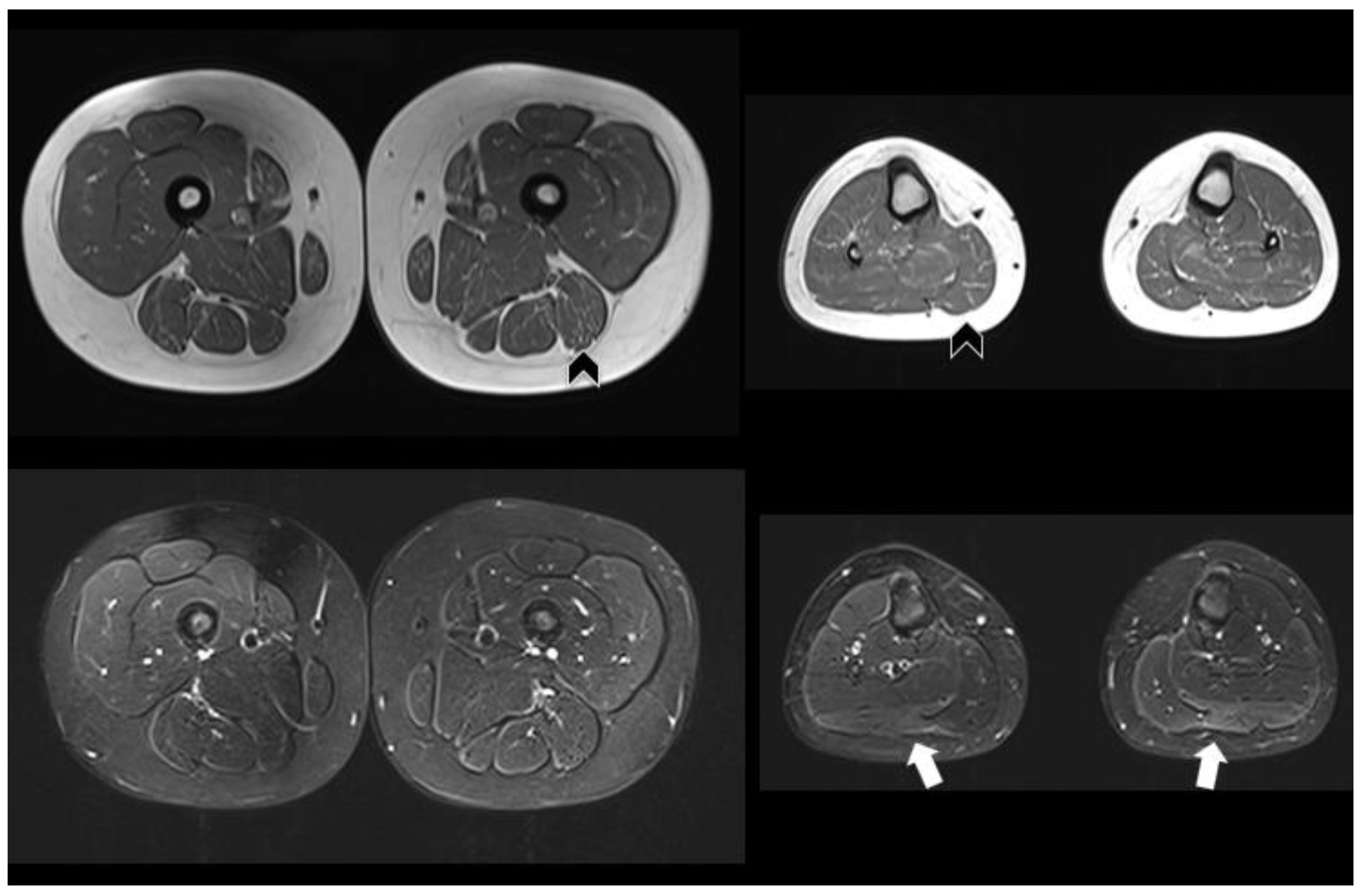

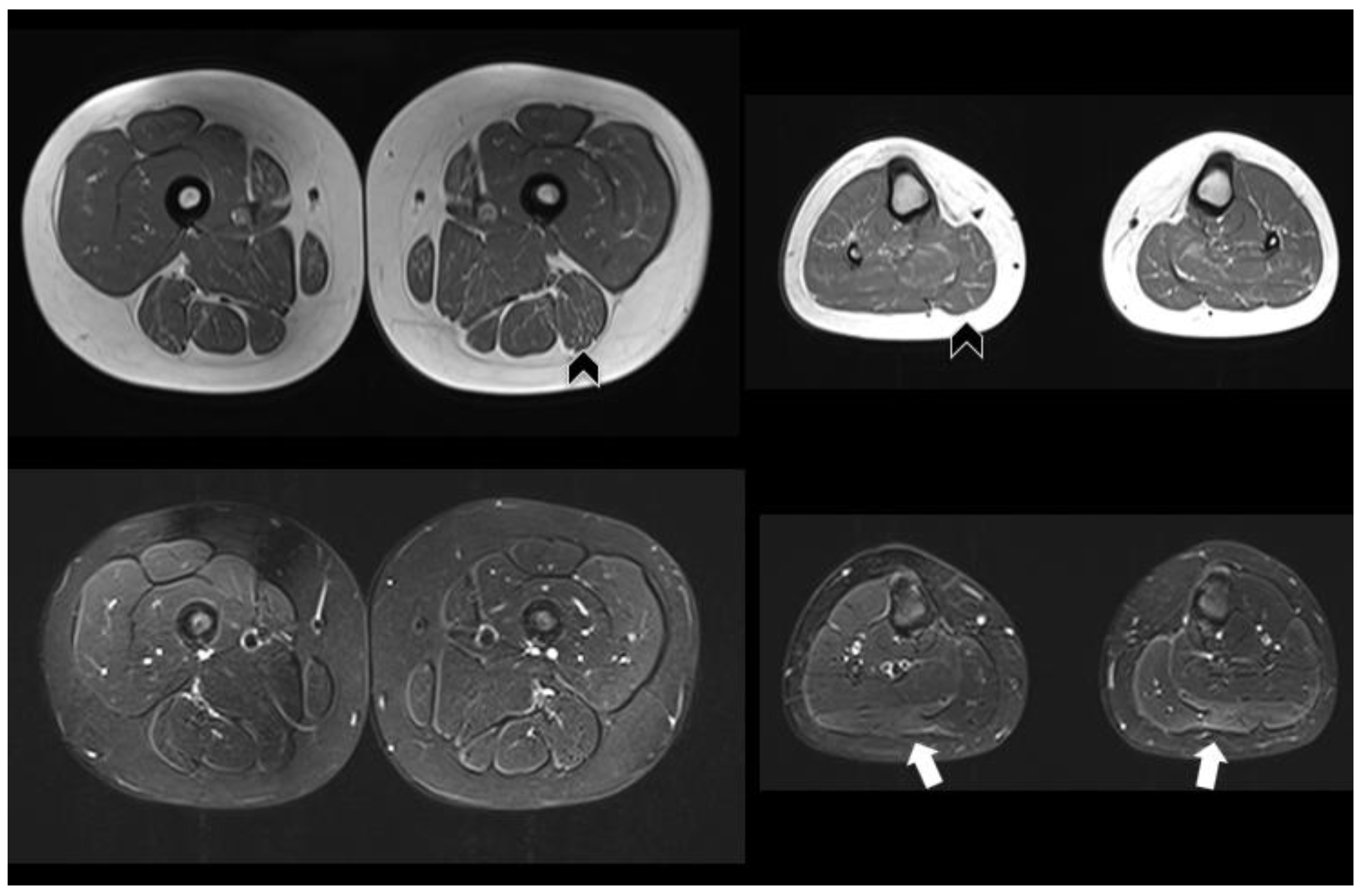{kind=link}
{kind=link}
| Patient 1 7 Months Old | Patient 2 8 Years Old | Patient 3 10 Years Old | Mother | |
|---|---|---|---|---|
| Symptoms | None | Slight fatigability in the previous four months | None | Muscle fatigability and cramps from a young age |
| Signs of disease are present at evaluation | Slight hypotonia of the upper back | Difficulty in whistling and blowing properly | None | None |
| Serum Creatine Phosphokinase (CPK, U/L) Normal value < 190 | 372 | 153 | 183 | 134 |
| Lactate Dehydrogenase (LDH, U/L) Normal value 120–300 | 342 | 357 | 242 | Not available |
| Aspartate Transaminase (AST, U/L) Normal value < 40 | 40 | 25 | 20 | Not available |
| Alanine Transaminase (ALT, U/L) Normal value < 41 | 24 | 17 | 25 | Not available |
| Creatine Kinase Muscle Band (CK-MB, ng/mL) Normal value < 6.22 | 11.8 | 3.41 | 3.3 | Not available |
| Pro-B-type Natriuretic Peptide (pro-BNP, pg/mL) Normal value < 320 | 31 | 79 | 52 | Not available |
| Acid maltase activity on leukocytes Normal value > 0.35 | 0.33 | Not performed | Not performed | Not performed |
| Enzyme activity on a dry blood spot (µmol/L/h) Normal value > 2 | 1.9 | 1.2 | 1.2 | |
| Urinary Tetrasaccharid (Glc4, mmol/mol/cr) Normal value 0.08–1.37 | 3.21 | 0.78 | 0.69 | Not performed |
| GAA sequencing | c.2284G>A p.(Glu762Lys); c.1994G>A p.(Gly665Glu) | c.2284G>A p.(Glu762Lys); c.1994G>A p.(Gly665Glu) | c.2284G>A p.(Glu762Lys); c.1994G>A p.(Gly665Glu) | c.1994G>A p.(Gly665Glu) |
| Electromyography | Normal | Normal | Normal | Normal |
| Heart ultrasound | Normal | Normal | Normal | Not performed |
| Spirometry | Not performed | Normal | Normal | Not performed |
| Six Minute Walking Test | Not performed | Normal | Normal | Not performed |
| Muscle MRI | Not performed | Pathologic (Figure 1) | Normal | Not performed |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faraguna, M.C.; Crescitelli, V.; Fornari, A.; Barzaghi, S.; Savasta, S.; Foiadelli, T.; Veraldi, D.; Paoletti, M.; Pichiecchio, A.; Gasperini, S. Treatment Dilemma in Children with Late-Onset Pompe Disease. Genes 2023, 14, 362. https://doi.org/10.3390/genes14020362
Faraguna MC, Crescitelli V, Fornari A, Barzaghi S, Savasta S, Foiadelli T, Veraldi D, Paoletti M, Pichiecchio A, Gasperini S. Treatment Dilemma in Children with Late-Onset Pompe Disease. Genes. 2023; 14(2):362. https://doi.org/10.3390/genes14020362
Chicago/Turabian StyleFaraguna, Martha Caterina, Viola Crescitelli, Anna Fornari, Silvia Barzaghi, Salvatore Savasta, Thomas Foiadelli, Daniele Veraldi, Matteo Paoletti, Anna Pichiecchio, and Serena Gasperini. 2023. "Treatment Dilemma in Children with Late-Onset Pompe Disease" Genes 14, no. 2: 362. https://doi.org/10.3390/genes14020362
APA StyleFaraguna, M. C., Crescitelli, V., Fornari, A., Barzaghi, S., Savasta, S., Foiadelli, T., Veraldi, D., Paoletti, M., Pichiecchio, A., & Gasperini, S. (2023). Treatment Dilemma in Children with Late-Onset Pompe Disease. Genes, 14(2), 362. https://doi.org/10.3390/genes14020362