
A Novel DLG1 Variant in a Family with Brugada Syndrome: Clinical Characteristics and In Silico Analysis


 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical and Genetic Investigation
2.2. Whole Exome Sequencing
2.3. Sanger Sequencing
2.4. In Silico Analysis and Protein Structure Modeling
3. Results
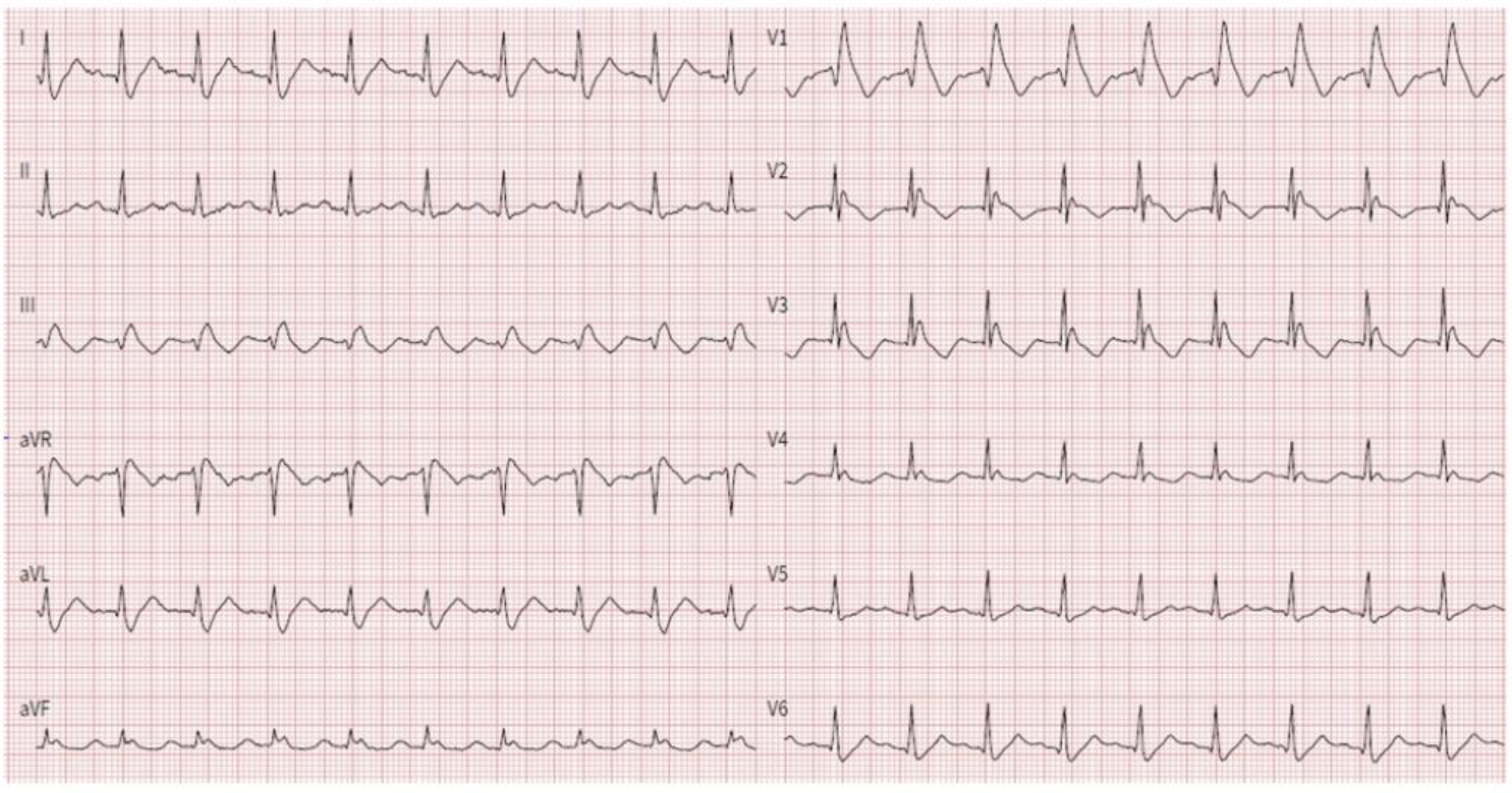
3.1. Proband Characteristics
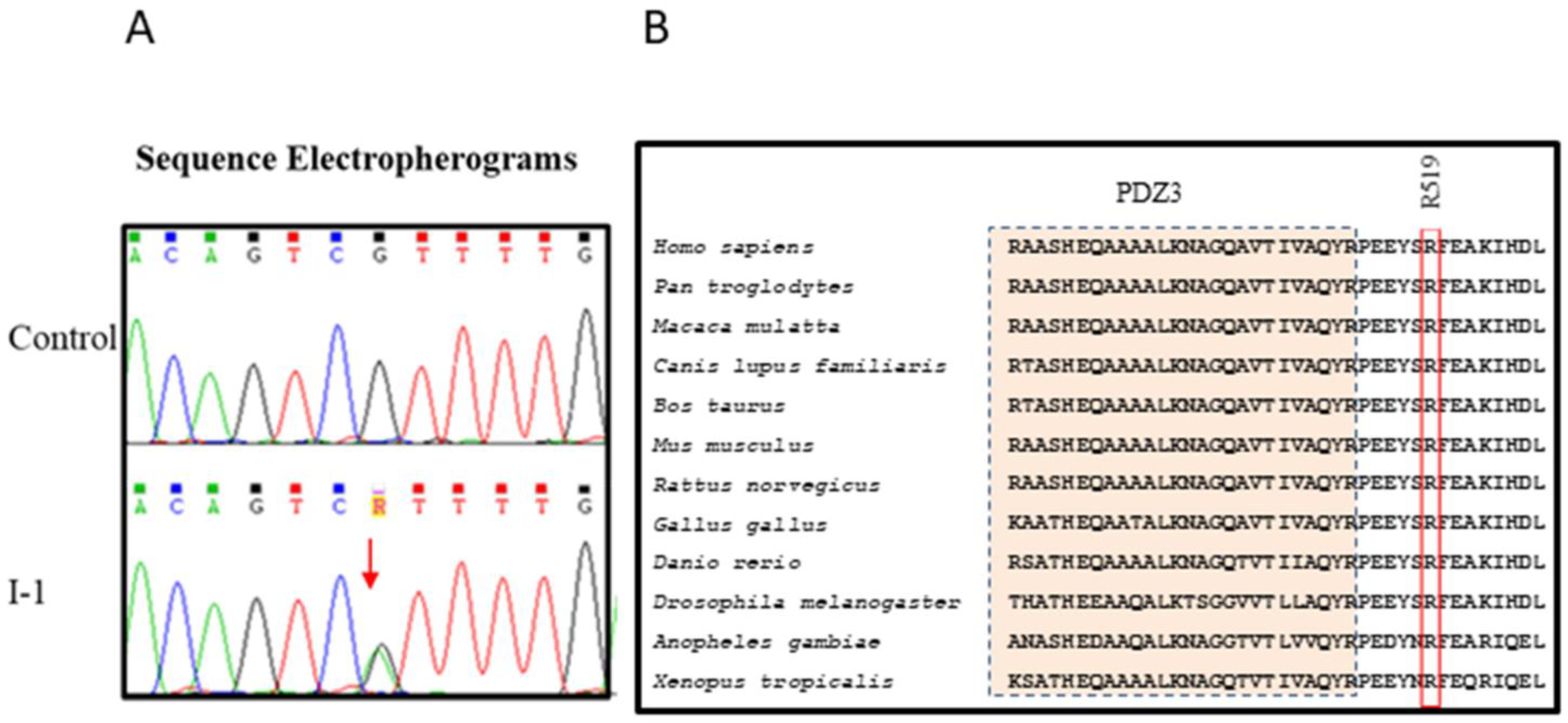
3.2. Whole-Exome Sequencing of Brugada Syndrome Patients Revealed Variant in Gene Known for Their Association with the Disease
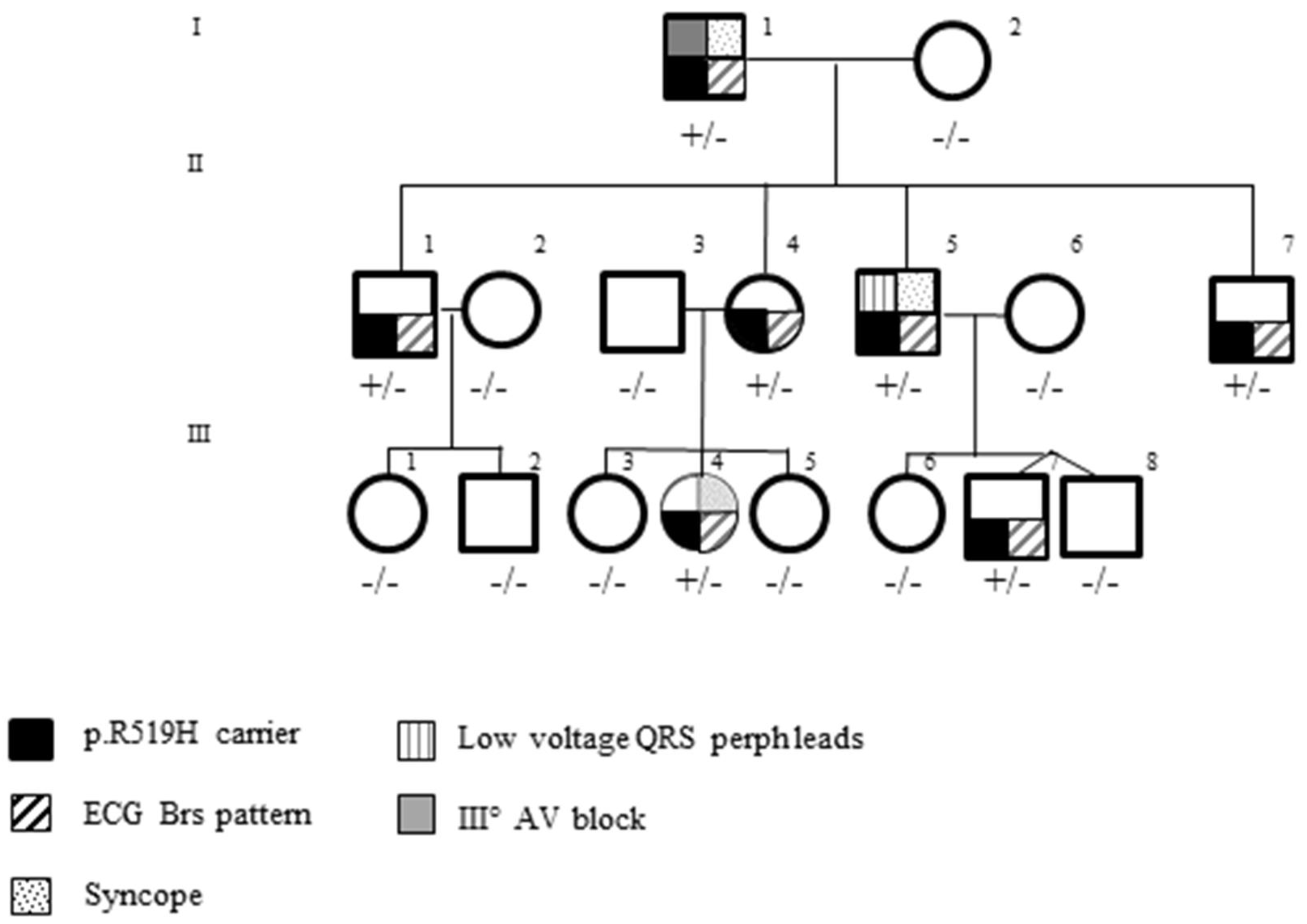
3.3. Pedigree Investigation
4. Discussion
4.1. Genes and BrS
4.2. Ion Channels and BrS
4.3. Ion Channel-Interacting Proteins and BrS
4.4. DLG1 Gene Encoding SAP97 Protein
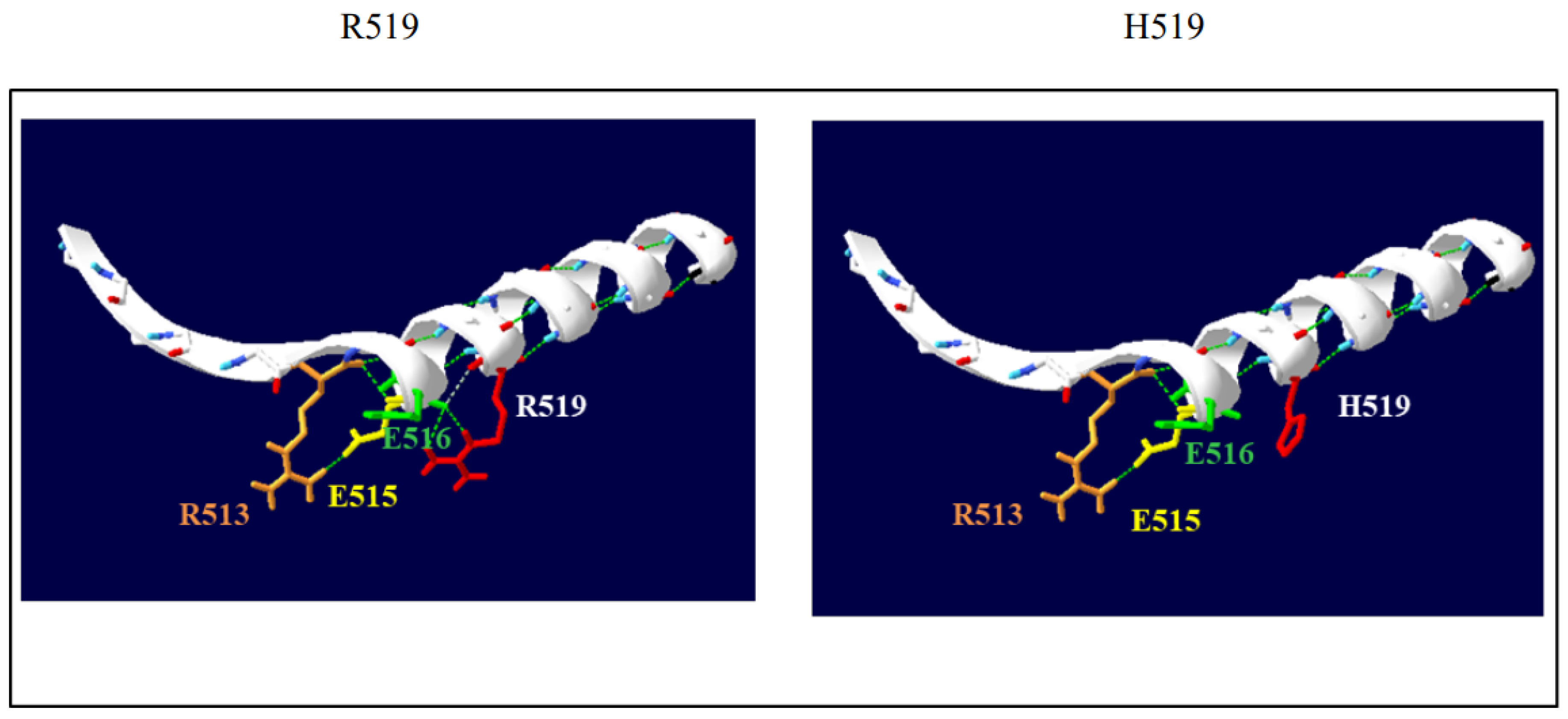
4.5. DLG1 Gene p.R519H Variant
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kobayashi, T.; Shintani, U.; Yamamoto, T.; Shida, S.; Isshiki, N.; Tanaka, T.; Ohmoto, Y.; Kitamura, M.; Kato, S.; Misaki, M. Familial Occurrence of Electrocardiographic Abnormalities of the Brugada-Type. Intern. Med. 1996, 35, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome JACC State-of-the-Art Review. JACC 2018, 72, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- ESC Scientific Document Group. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.; Postema, P.G.; Di Diego, J.M.; Viskin, S.; Morita, H.; Fish, J.M. The pathophysiological mechanism underlying Brugada syndrome: Depolarization versus repolarization. J. Mol. Cell. Cardiol. 2010, 49, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Tse, G.; Wong, S.T.; Tse, V.; Yeo, J.M. Depolarization vs. repolarization: What is the mechanism of ventricular arrhythmogenesis underlying sodium channel haploinsufficiency in mouse hearts? Acta Physiol. 2016, 218, 236–238. [Google Scholar] [CrossRef]
- Grant, A.O. Cardiac Ion Channels. Circ. Arrhythmia Electrophysiol. 2009, 2, 185–194. [Google Scholar] [CrossRef]
- Sarquella-Brugada, G.; Campuzano, O.; Arbelo, E.; Brugada, J.; Brugada, R. Brugada syndrome: Clinical and genetic findings. Genet. Med. 2016, 18, 3–12. [Google Scholar] [CrossRef]
- Gourraud, J.-B.; Barc, J.; Thollet, A.; Le Scouarnec, S.; Le Marec, H.; Schott, J.-J.; Redon, R.; Probst, V. The Brugada Syndrome: A Rare Arrhythmia Disorder with Complex Inheritance. Front. Cardiovasc. Med. 2016, 3, 9. [Google Scholar] [CrossRef]
- Probst, V.; Wilde, A.A.; Barc, J.; Sacher, F.; Babuty, D.; Mabo, P.; Mansourati, J.; Le Scouarnec, S.; Kyndt, F.; Le Caignec, C.; et al. SCN5A Mutations and the Role of Genetic Background in the Pathophysiology of Brugada Syndrome. Circ. Cardiovasc. Genet. 2009, 2, 552–557. [Google Scholar] [CrossRef]
- Marian, A.J. Nature’s Genetic Gradients and the Clinical Phenotype. Circ. Cardiovasc. Genet. 2009, 2, 537–539. [Google Scholar] [CrossRef]
- Hosseini, S.M.; Kim, R.; Udupa, S.; Costain, G.; Jobling, R.; Liston, E.; Jamal, S.M.; Szybowska, M.; Morel, C.F.; Bowdin, S.; et al. Reappraisal of reported genes for sudden arrhythmic death. Circulation 2018, 138, 1195–1205. [Google Scholar] [CrossRef]
- Kapplinger, J.D.; Tester, D.J.; Alders, M.; Benito, B.; Berthet, M.; Brugada, J.; Brugada, P.; Fressart, V.; Guerchicoff, A.; Harris-Kerr, C.; et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm 2010, 7, 33–46. [Google Scholar] [CrossRef]
- Di Resta, C.; Berg, J.; Villatore, A.; Maia, M.; Pili, G.; Fioravanti, F.; Tomaiuolo, R.; Sala, S.; Benedetti, S.; Peretto, G. Concealed Substrates in Brugada Syndrome: Isolated Channelopathy or Associated Cardiomyopathy? Genes 2022, 13, 1755. [Google Scholar] [CrossRef]
- Balse, E.; Boycott, H.E. Ion channel trafficking: Control of ion channel density as a target for arrhythmias? Front. Physiol. 2017, 8, 808. [Google Scholar] [CrossRef]
- Fourie, C.; Li, D.; Montgomery, J.M. The anchoring protein SAP97 influences the trafficking and localisation of multiple membrane channels. Biochim. Biophys. Acta 2014, 1838, 589–594. [Google Scholar] [CrossRef]
- Abi-Char, J.; El-Haou, S.; Balse, E.; Neyroud, N.; Vranckx, R.; Coulombe, A.; Hatem, S.N. The anchoring protein SAP97 retains Kv1.5 channels in the plasma membrane of cardiac myocytes. Am. J. Physiol. Circ. Physiol. 2008, 294, H1851–H1861. [Google Scholar] [CrossRef]
- Godreau, D.; Vranckx, R.; Maguy, A.; Rücker-Martin, C.; Goyenvalle, C.; Abdelshafy, S.; Tessier, S.; Couétil, J.P.; Hatem, S.N. Expression, regulation and role of the MAGUK protein SAP-97 in human atrial myocardium. Cardiovasc. Res. 2002, 56, 433–442. [Google Scholar] [CrossRef]
- El-Haou, S.; Balse, E.; Neyroud, N.; Dilanian, G.; Gavillet, B.; Abriel, H.; Coulombe, A.; Jeromin, A.; Hatem, S.N. Kv4 Potassium Channels Form a Tripartite Complex With the Anchoring Protein SAP97 and CaMKII in Cardiac Myocytes. Circ. Res. 2009, 104, 758–769. [Google Scholar] [CrossRef]
- Gillet, L.; Rougier, J.S.; Shy, D.; Sonntag, S.; Mougenot, N.; Essers MShmerling, D.; Balse, E.; Hatem, S.N.; Abriel, H. Cardiac Specific Ablation of Synapse-Associated Protein SAP97 in Mice Decreases Potassium Currents but Not Sodium Current. Heart Rhythm 2015, 12, 181–192. [Google Scholar] [CrossRef]
- Leonoudakis, D.; Mailliard, W.; Wingerd, K.; Clegg, D.; Vandenberg, C. Inward rectifier potassium channel Kir2.2 is associated with synapse-associated protein SAP97. J. Cell Sci. 2001, 114, 987–998. [Google Scholar] [CrossRef]
- Milstein, M.L.; Musa, H.; Balbuena, D.P.; Anumonwo, J.M.B.; Auerbach, D.S.; Furspan, P.B.; Hou, L.; Hu, B.; Schumacher, S.M.; Vaidyanathan, R.; et al. Dynamic reciprocity of sodium and potassium channel expression in a macromolecular complex controls cardiac excitability and arrhythmia. Proc. Natl. Acad. Sci. USA 2012, 109, E2134–E2143. [Google Scholar] [CrossRef] [PubMed]
- Matamoros, M.; Pérez-Hernández, M.; Guerrero-Serna, G.; Amorós, I.; Barana, A.; Núñez, M.; Ponce-Balbuena, D.; Sacristán, S.; Gómez, R.; Tamargo, J.; et al. Nav1.5 N-terminal Domain Binding to A1-Syntrophin Increases Membrane Density of Human Kir2.1, Kir2.2 and Nav1.5 Channels. Cardiovasc. Res. 2016, 110, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, S.; Zmoos, A.F.; Ogrodnik, J.; Balse, E.; Raad, N.; El-Haou, S.; Albesa, M.; Bittihn, P.; Luther, S.; Lehnart, S.E.; et al. SAP97 and Dystrophin Macromolecular Complexes Determine Two Pools of Cardiac Sodium Channels Nav1.5 in Cardiomyocytes. Circ. Res. 2011, 108, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, R.; Taffet, S.M.; Vikstrom, K.L.; Anumonwo, J.M.B. Regulation of Cardiac Inward Rectifier Potassium Current (IK1) by Synapse-associated Protein-97. J. Biol. Chem. 2010, 285, 28000–28009. [Google Scholar] [CrossRef] [PubMed]
- Musa, H.; Marcou, C.A.; Herron, T.J.; Makara, M.A.; Tester, D.J.; O’Connell, R.P.; Rosinski, B.; Guerrero-Serna, G.; Milstein, M.L.; Da Rocha, A.M.; et al. Abnormal myocardial expression of SAP97 is associated with arrhythmogenic risk. Am. J. Physiol. Circ. Physiol. 2020, 318, H1357–H1370. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Antzelevitch, C.; Borggrefe, M.; Brugada, J.; Brugada, R.; Brugada, P.; Corrado, D.; Hauer, R.N.W.; Kass, R.S.; Nademanee, K.; et al. Study Group on the Molecular Basis of Arrhythmias of the European Society of Cardiology Proposed diagnostic criteria for the Brugada syndrome: Consensus report. Circulation 2002, 106, 2514–2519. [Google Scholar] [CrossRef]
- Therasse, D.; Sacher, F.; Petit, B.; Babuty, D.; Mabo, P.; Martins, R.; Jesel, L.; Maury, P.; Pasquie, J.L.; Mansourati, J.; et al. Sodium-channel blocker challenge in the familial screening of Brugada syndrome: Safety and predictors of positivity. Heart Rhythm 2017, 14, 1442–1448. [Google Scholar] [CrossRef]
- Sue, R.; Nazneen, A.; Sherri, B.; David, B.; Soma, D.; Gastier-Foster, J.; Grody, W.W.; Madhuri, H.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar]
- Ariano, A.; D’Apolito, M.; Bova, M.; Bellanti, F.; Loffredo, S.; D’Andrea, G.; Intrieri, M.; Petraroli, A.; Maffione, A.B.; Spadaro, G.; et al. A myoferlin gain-of-function variant associates with a new type of hereditary angioedema. Allergy 2020, 75, 2989–2992. [Google Scholar] [CrossRef]
- D’Andrea, G.; Colaizzo, D.; Vecchione, G.; Grandone, E.; Di Minno, G.; Margaglione, M.; the GLAnzmann’s Thrombasthenia Italian Team (GLATIT). Glanzmann’s Thrombasthenia: Identification of 19 New Mutations in 30 Patients. Thromb. Haemost. 2002, 87, 1034–1042. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Giordano, U.; Collisani, G.; Memmi, M. Brugada syndrome and sudden cardiac death in children. Lancet 2000, 355, 808–809. [Google Scholar] [CrossRef]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.-B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. [Google Scholar] [CrossRef]
- Monasky, M.M.; Micaglio, E.; Ciconte, G.; Pappone, C. Brugada syndrome: Oligogenic or mendelian disease? Int. J. Mol. Sci. 2020, 21, 1687. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Ye, D.; Tester, D.J.; Crotti, L.; Mugione, A.; Nesterenko, V.V.; Albertson, R.M.; Antzelevitch, C.; Schwartz, P.J.; Ackerman, M.J. Transient outward current (I(to)) gain-of-function mutations in the KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart Rhythm 2011, 8, 1024–1032. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Ackerman, M.J. Potassium-channel mutations and cardiac arrhythmias--diagnosis and therapy. Rev. Cardiol. 2012, 9, 319–332. [Google Scholar] [CrossRef]
- Kattygnarath, D.; Maugenre, S.; Neyroud, N.; Balse, E.; Ichai, C.; Denjoy, I.; Dilanian, G.; Martins, R.P.; Fressart, V.; Berthet, M.; et al. MOG1: A new susceptibility gene for Brugada syndrome. Circ. Cardiovasc. Genet. 2011, 4, 261–268. [Google Scholar] [CrossRef]
- Delpon, E.; Tinaquero, D.; Alfayate, S.; Nieto-Marin, P.; Utrilla, R.; Matamoros, M.; Perez-Hernandez, M.; Tamargo, M.; Toquero, J.; Cosio, F.; et al. P6281A DLG1 polymorphism shortens the action potential duration and the QT interval. Eur. Heart J. 2017, 38 (Suppl. S1), ehx493. [Google Scholar] [CrossRef]
- Tinaquero, D.; Crespo-García, T.; Utrilla, R.G.; Nieto-Marín, P.; González-Guerra, A.; Rubio-Alarcón, M.; Cámara-Checa, A.; Dago, M.; Matamoros, M.; Pérez-Hernández, M.; et al. The p.P888L SAP97 polymorphism increases the transient outward current (Ito,f) and abbreviates the action potential duration and the QT interval. Sci. Rep. 2020, 10, 10707. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Nucleotide Change | Amino Acid Change | PolyPhen-2 | SIFT | CADD |
|---|---|---|---|---|---|
| DGL1 | CGT/CAT | R519H | 1 | 0 | 31 |
| Patient | Sex | Age | History of Syncope | ECG Features | Echocardiogram Features |
|---|---|---|---|---|---|
| I | M | 74 | Yes | Spontaneous type 1 BrS pattern; III AVB | Impaired LV-GLS; Sigmoid-shaped IV septum |
| II-1 | M | 54 | No | Type 2 BrS pattern | -- |
| II-4 | F | 48 | No | Type 2 BrS pattern | -- |
| II-5 | M | 47 | Yes | Type 2 BrS pattern Low-voltage peripheric limbs | Impaired LV-GLS |
| II-6 | M | 43 | No | Type 2 BrS pattern | -- |
| III-4 | F | 20 | Yes | Type 2 BrS pattern | Mitral valve prolapse |
| III-7 | M | 11 | No | Type 2 BrS pattern | -- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
d’Apolito, M.; Santoro, F.; Santacroce, R.; Cordisco, G.; Ragnatela, I.; D’Arienzo, G.; Pellegrino, P.L.; Brunetti, N.D.; Margaglione, M. A Novel DLG1 Variant in a Family with Brugada Syndrome: Clinical Characteristics and In Silico Analysis. Genes 2023, 14, 427. https://doi.org/10.3390/genes14020427
d’Apolito M, Santoro F, Santacroce R, Cordisco G, Ragnatela I, D’Arienzo G, Pellegrino PL, Brunetti ND, Margaglione M. A Novel DLG1 Variant in a Family with Brugada Syndrome: Clinical Characteristics and In Silico Analysis. Genes. 2023; 14(2):427. https://doi.org/10.3390/genes14020427
Chicago/Turabian Styled’Apolito, Maria, Francesco Santoro, Rosa Santacroce, Giorgia Cordisco, Ilaria Ragnatela, Girolamo D’Arienzo, Pier Luigi Pellegrino, Natale Daniele Brunetti, and Maurizio Margaglione. 2023. "A Novel DLG1 Variant in a Family with Brugada Syndrome: Clinical Characteristics and In Silico Analysis" Genes 14, no. 2: 427. https://doi.org/10.3390/genes14020427
APA Styled’Apolito, M., Santoro, F., Santacroce, R., Cordisco, G., Ragnatela, I., D’Arienzo, G., Pellegrino, P. L., Brunetti, N. D., & Margaglione, M. (2023). A Novel DLG1 Variant in a Family with Brugada Syndrome: Clinical Characteristics and In Silico Analysis. Genes, 14(2), 427. https://doi.org/10.3390/genes14020427