
Spotlight on hTERT Complex Regulation in Cutaneous T-Cell Lymphomas
 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Methods
2.1. Patients and Healthy Controls
2.2. CTCL Cell Lines
2.3. Telomerase Activity Estimation
2.4. Nucleic Acid Isolation
2.5. Telomere Length Measurement
2.6. hTERT Hotspot Promoter Mutations Detection
2.7. hTERT SNPs Genotyping
2.8. hTERT Splicing Variants Expression
2.9. Lentiviral shRNA Construction and Production
2.10. Lentiviral Cell Transduction
2.11. Cell Proliferation Analysis
2.12. Cell Clonogenicity Analysis
2.13. Statistical Analysis
3. Results
3.1. hTERT SNPs Are Associated with CTCL Risk in Patient Cells
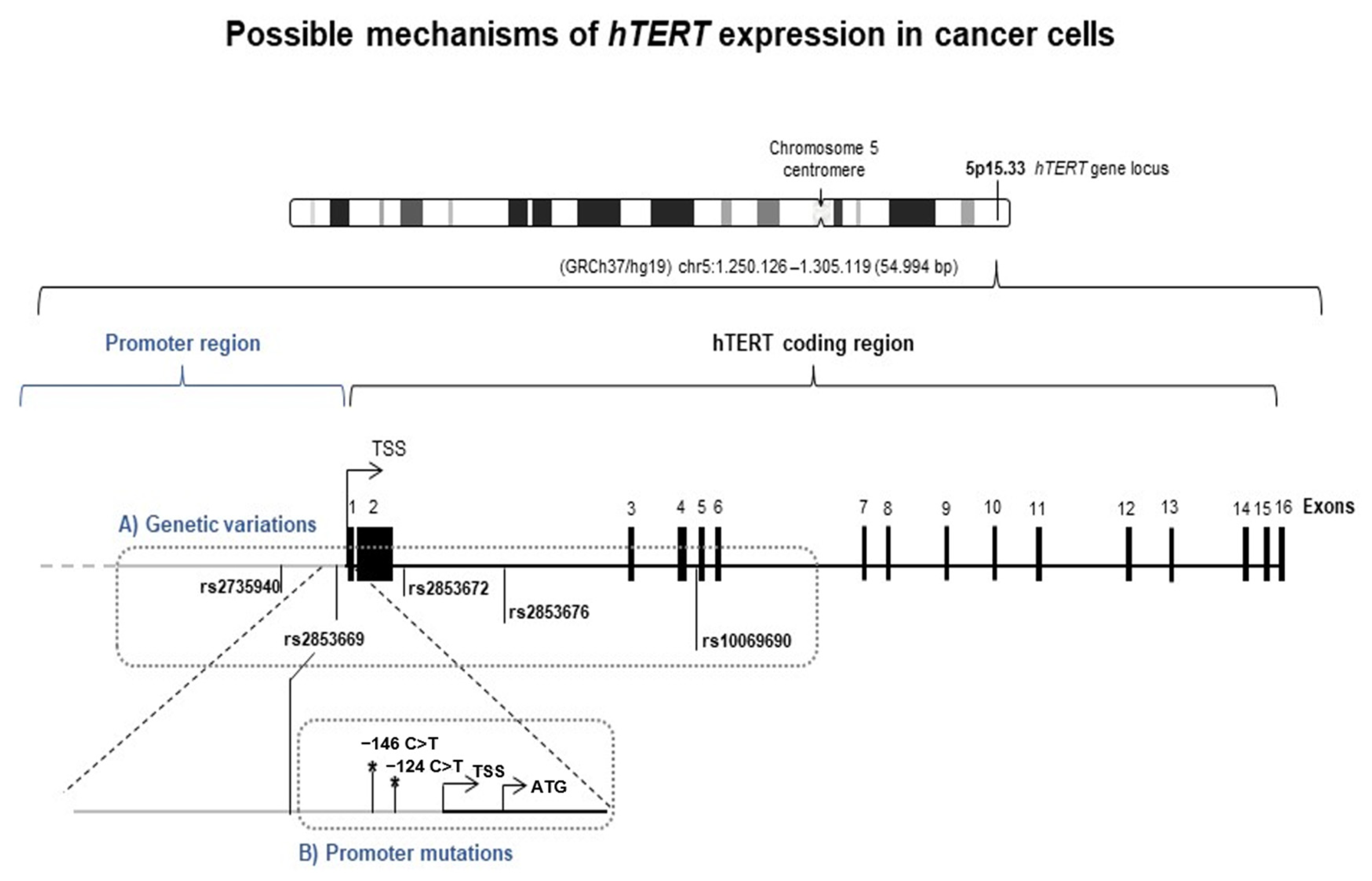
3.2. hTERT Transcription Regulation in CTCL: hTERT Promoter Mutations Are a Rare Event in CTCL Patients
3.3. hTERT Post-Transcription Regulation in CTCL Cells
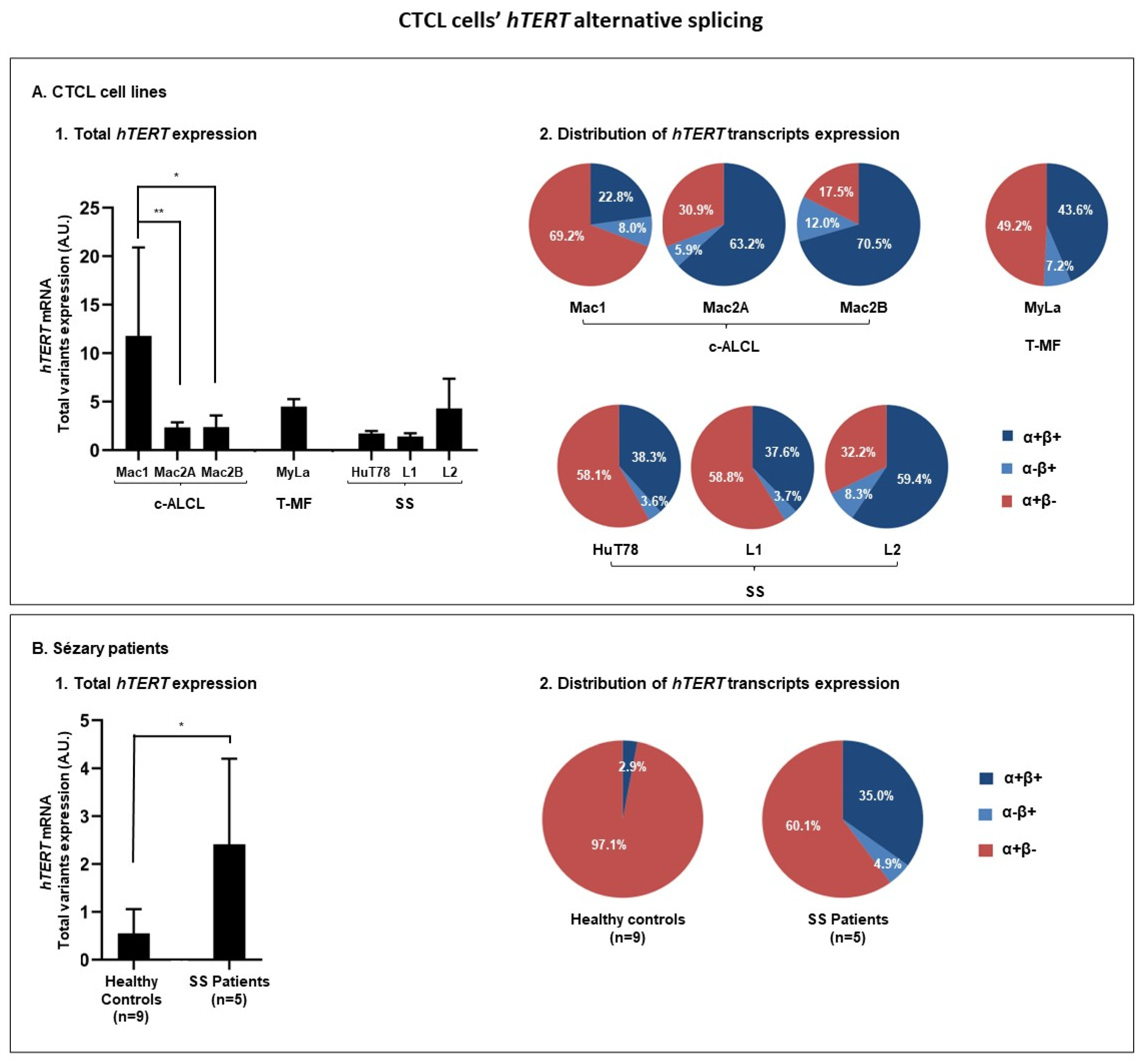
3.3.1. hTERT Is Subjected to Alternative Splicing in CTCL Cells
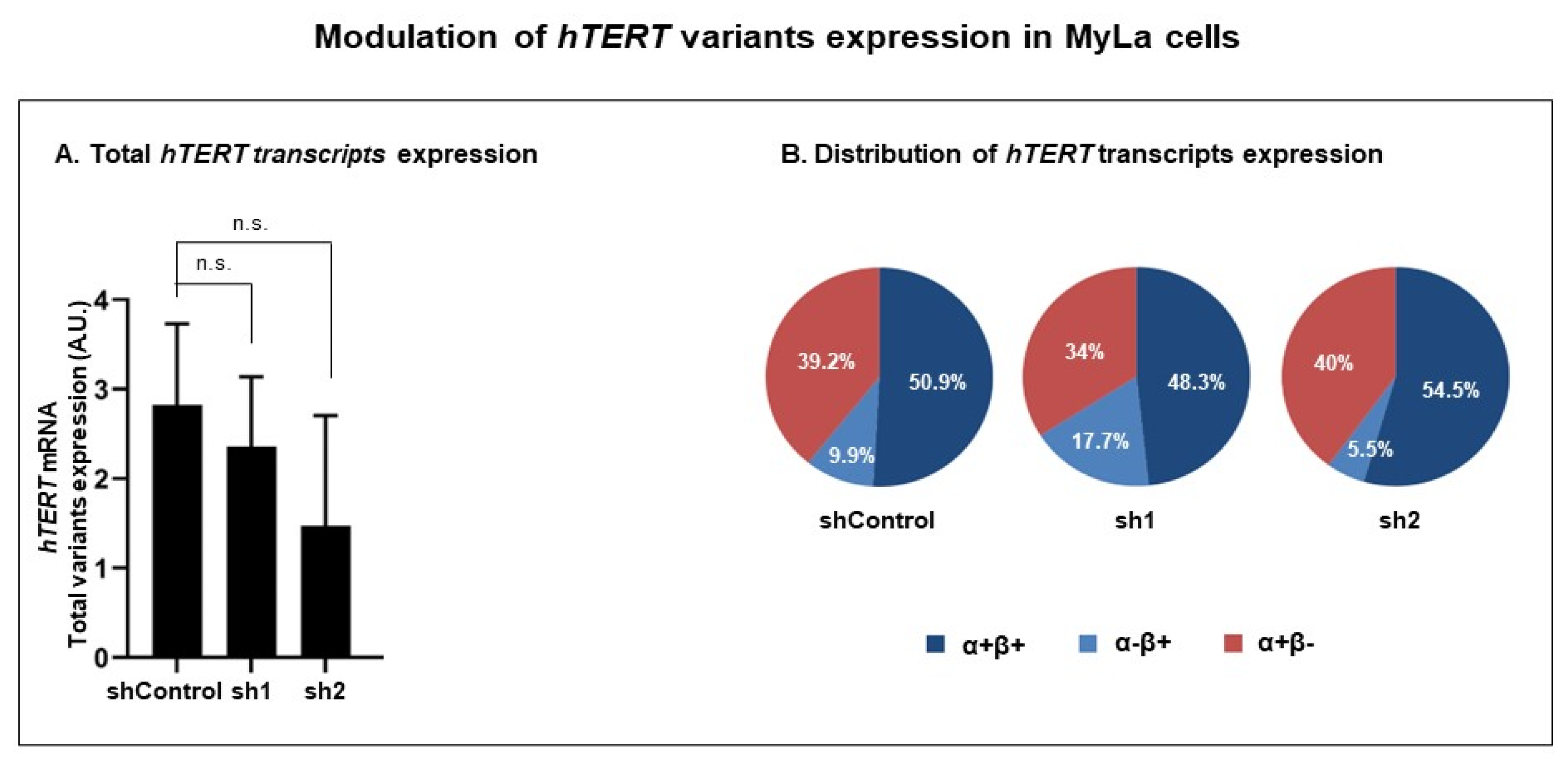
3.3.2. Modulation of RT Domain Transcriptome
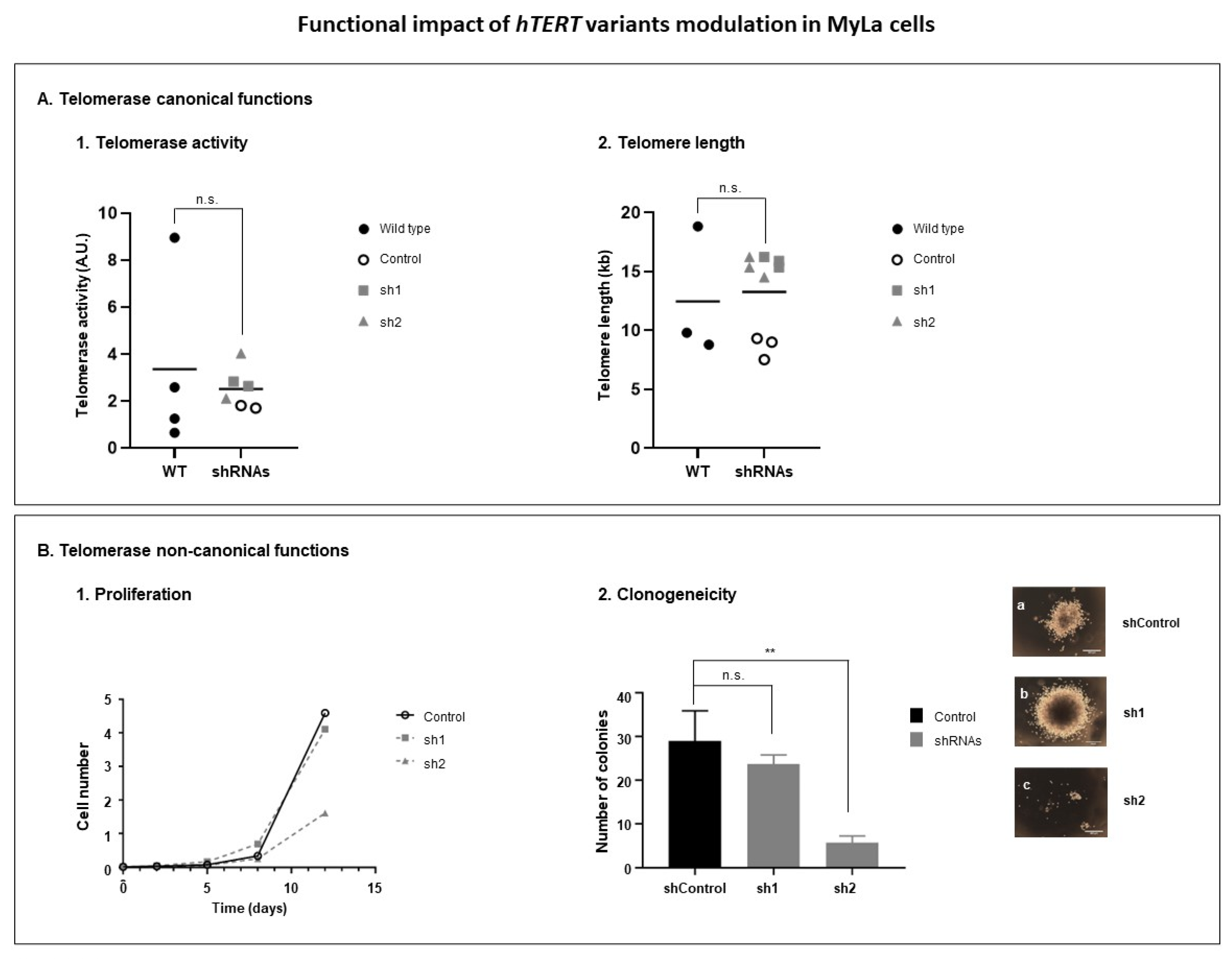
3.3.3. Modulation of hTERT Transcriptome Affects Telomerase Non-Canonical Functions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hinds, P.; Pietruska, J.J.F. Senescence and tumor suppression. F1000Research 2017, 6, 2121. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, T.B.; Sá, A.; Lopes, J.M.; Sobrinho-Simões, M.; Soares, P.; Vinagre, J. Telomere maintenance mechanisms in cancer. Genes 2018, 9, 241. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H.; Collins, K. Telomerase: An RNP enzyme synthesizes DNA. Cold Spring Harb. Perspect. Biol. 2011, 3, a003558. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Snow, B.; Hande, M.; Yeung, D.; Erdmann, N.; Wakeham, A.; Itie, A.; Siderovski, D.; Lansdorp, P.; Robinson, M. The telomerase reverse transcriptase is limiting and necessary for telomerase function in vivo. Curr. Biol. 2000, 10, 1459–1462. [Google Scholar] [CrossRef] [PubMed]
- Penna, G.C.; Pestana, A.; Cameselle, J.M.; Momesso, D.; de Andrade, F.A.; Vidal, A.P.A.; Junior, M.L.A.; Melo, M.; Fernandes, P.V.; Corbo, R. TERTp mutation is associated with a shorter progression free survival in patients with aggressive histology subtypes of follicular-cell derived thyroid carcinoma. Endocrine 2018, 61, 489–498. [Google Scholar] [PubMed]
- Low, K.C.; Tergaonkar, V. Telomerase: Central regulator of all of the hallmarks of cancer. Trends Biochem. Sci. 2013, 38, 426–434. [Google Scholar]
- Ding, D.; Zhou, J.; Wang, M.; Cong, Y.S. Implications of telomere-independent activities of telomerase reverse transcriptase in human cancer. FEBS J. 2013, 280, 3205–3211. [Google Scholar]
- Pestana, A.; Vinagre, J.; Sobrinho-Simões, M.; Soares, P. TERT biology and function in cancer: Beyond immortalisation. J. Mol. Endocrinol. 2017, 58, R129–R146. [Google Scholar] [CrossRef]
- Leão, R.; Apolónio, J.D.; Lee, D.; Figueiredo, A.; Tabori, U.; Castelo-Branco, P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: Clinical impacts in cancer. J. Biomed. Sci. 2018, 25, 22. [Google Scholar]
- Gladych, M.; Wojtyla, A.; Rubis, B. Human telomerase expression regulation. Biochem. Cell Biol. 2011, 89, 359–376. [Google Scholar]
- Plyasova, A.A.; Zhdanov, D.D. Alternative Splicing of Human Telomerase Reverse Transcriptase (hTERT) and Its Implications in Physiological and Pathological Processes. Biomedicines 2021, 9, 526. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [PubMed]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [PubMed]
- Heidenreich, B.; Rachakonda, P.S.; Hemminki, K.; Kumar, R. TERT promoter mutations in cancer development. Curr. Opin. Genet. Dev. 2014, 24, 30–37. [Google Scholar] [PubMed]
- Vinagre, J.; Almeida, A.; Pópulo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 2185. [Google Scholar]
- Bournaud, C.; Descotes, F.; Decaussin-Petrucci, M.; Berthiller, J.; de la Fouchardière, C.; Giraudet, A.-L.; Bertholon-Gregoire, M.; Robinson, P.; Lifante, J.-C.; Lopez, J.; et al. TERT promoter mutations identify a high-risk group in metastasis-free advanced thyroid carcinoma. Eur. J. Cancer 2019, 108, 41–49. [Google Scholar] [PubMed]
- Ulaner, G.A.; Hu, J.F.; Vu, T.H.; Giudice, L.C.; Hoffman, A.R. Tissue-specific alternate splicing of human telomerase reverse transcriptase (hTERT) influences telomere lengths during human development. Int. J. Cancer 2001, 91, 644–649. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Chang, G.; Wang, F.; Wang, F.; Geng, X. Alternative splicing of hTERT Pre-mRNA: A potential strategy for the regulation of telomerase activity. Int. J. Mol. Sci. 2017, 18, 567. [Google Scholar] [CrossRef]
- Yi, X.; Shay, J.W.; Wright, W.E. Quantitation of telomerase components and hTERT mRNA splicing patterns in immortal human cells. Nucleic Acids Res. 2001, 29, 4818–4825. [Google Scholar]
- Lincz, L.F.; Mudge, L.-M.; Scorgie, F.E.; Sakoff, J.A.; Hamilton, C.S.; Seldon, M. Quantification of hTERT splice variants in melanoma by SYBR green real-time polymerase chain reaction indicates a negative regulatory role for the β deletion variant. Neoplasia 2008, 10, 1131. [Google Scholar]
- Zhdanov, D.; Vasina, D.; Orlova, V.; Gotovtseva, V.; Bibikova, M.; Pokrovsky, V.; Pokrovskayaa, M.; Aleksandrova, S.; Sokolov, N. Apoptotic endonuclease EndoG induces alternative splicing of telomerase catalytic subunit hTERT and death of tumor cells. Biochem. Suppl. Ser. B Biomed. Chem. 2016, 10, 310–321. [Google Scholar]
- Yi, X.; White, D.M.; Aisner, D.L.; Baur, J.A.; Wright, W.E.; Shay, J.W. An alternate splicing variant of the human telomerase catalytic subunit inhibits telomerase activity. Neoplasia 2000, 2, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Sæbøe-Larssen, S.; Fossberg, E.; Gaudernack, G. Characterization of novel alternative splicing sites in human telomerase reverse transcriptase (hTERT): Analysis of expression and mutual correlation in mRNA isoforms from normal and tumour tissues. BMC Mol. Biol. 2006, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Colgin, L.M.; Wilkinson, C.; Englezou, A.; Kilian, A.; Robinson, M.O.; Redder, R.R. The hTERTα splice variant is a dominant negative inhibitor of telomerase activity. Neoplasia 2000, 2, 426. [Google Scholar] [PubMed]
- Listerman, I.; Sun, J.; Gazzaniga, F.S.; Lukas, J.L.; Blackburn, E.H. The major reverse transcriptase–incompetent splice variant of the human telomerase protein inhibits telomerase activity but protects from apoptosis. Cancer Res. 2013, 73, 2817–2828. [Google Scholar] [CrossRef] [PubMed]
- Jie, M.-M.; Chang, X.; Zeng, S.; Liu, C.; Liao, G.-B.; Wu, Y.-R.; Liu, C.-H.; Hu, C.-J.; Yang, S.-M.; Li, X.-Z. Diverse regulatory manners of human telomerase reverse transcriptase. Cell Commun. Signal. 2019, 17, 63. [Google Scholar] [PubMed]
- Baird, D.M. Variation at the TERT locus and predisposition for cancer. Expert Rev. Mol. Med. 2010, 12, e16. [Google Scholar]
- Yang, H.; Li, J.; Tang, R.; Liu, Y.; Shao, Y.; Huang, Q.; Shi, J. Telomere reverse transcriptase (TERT) rs2735940 increases cancer risk. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2015, 21, 612. [Google Scholar]
- Matsubara, Y.; Murata, M.; Yoshida, T.; Watanabe, K.; Saito, I.; Miyaki, K.; Omae, K.; Ikeda, Y. Telomere length of normal leukocytes is affected by a functional polymorphism of hTERT. Biochem. Biophys. Res. Commun. 2006, 341, 128–131. [Google Scholar]
- Shen, N.; Lu, Y.; Wang, X.; Peng, J.; Zhu, Y.; Cheng, L. Association between rs2853669 in TERT gene and the risk and prognosis of human cancer: A systematic review and meta-analysis. Oncotarget 2017, 8, 50864. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, T.; Wu, Z.; Zhang, K.; Li, W.; Yang, J.; Chen, C.; Chen, L.; Xing, J. Association between TERT rs2853669 polymorphism and cancer risk: A meta-analysis of 9,157 cases and 11,073 controls. PLoS ONE 2018, 13, e0191560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Thakur, A.; Liang, Y.; Zhang, S.; Wang, T.; Chen, T.; Meng, J.; Wang, L.; Wu, F.; Jin, T. Polymorphisms in the TERT gene are associated with lung cancer risk in the Chinese Han population. Eur. J. Cancer Prev. 2014, 23, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.-L.; Yuan, P.; Abuduwufuer, A.; Lv, W.; Yang, Y.-H.; Hu, J. Association between the TERT genetic polymorphism rs2853676 and cancer risk: Meta-analysis of 76 108 cases and 134 215 controls. PLoS ONE 2015, 10, e0128829. [Google Scholar] [CrossRef]
- Matsubara, Y.; Murata, M.; Watanabe, K.; Saito, I.; Miyaki, K.; Omae, K.; Ishikawa, M.; Matsushita, K.; Iwanaga, S.; Ogawa, S. Coronary artery disease and a functional polymorphism of hTERT. Biochem. Biophys. Res. Commun. 2006, 348, 669–672. [Google Scholar] [CrossRef]
- Batista, R.; Cruvinel-Carloni, A.; Vinagre, J.; Peixoto, J.; Catarino, T.A.; Campanella, N.C.; Becker, A.P.; de Almeida, G.C.; Matsushita, M.M.; Clara, C.; et al. The prognostic impact of TERT promoter mutations in glioblastomas is modified by the rs2853669 single nucleotide polymorphism. Int. J. Cancer 2016, 139, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Helbig, S.; Wockner, L.; Bouendeu, A.; Hille-Betz, U.; McCue, K.; French, J.D.; Edwards, S.L.; Pickett, H.A.; Reddel, R.R.; Chenevix-Trench, G.; et al. Functional dissection of breast cancer risk-associated TERT promoter variants. Oncotarget 2017, 8, 67203. [Google Scholar]
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood J. Am. Soc. Hematol. 2019, 133, 1703–1714. [Google Scholar]
- Kamijo, H.; Miyagaki, T. Mycosis fungoides and Sézary syndrome: Updates and review of current therapy. Curr. Treat. Options Oncol. 2021, 22, 10. [Google Scholar] [CrossRef]
- Sethi, T.K.; Montanari, F.; Foss, F.; Reddy, N. How we treat advanced stage cutaneous T-cell lymphoma–mycosis fungoides and Sézary syndrome. Br. J. Haematol. 2021, 195, 352–364. [Google Scholar]
- Vural, S.; Akay, B.N.; Botsalı, A.; Atilla, E.; Parlak, N.; Heper, A.O.; Şanlı, H. Transformation of mycosis fungoides/Sezary syndrome: Clinical characteristics and prognosis. Turk. J. Hematol. 2018, 35, 35. [Google Scholar]
- Cristofoletti, C.; Narducci, M.G.; Russo, G.J.C.C.O. Sézary Syndrome, recent biomarkers and new drugs. Chin Clin Oncol 2019, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- García-Díaz, N.; Piris, M.Á.; Ortiz-Romero, P.L.; Vaqué, J.P. Mycosis fungoides and sézary syndrome: An integrative review of the pathophysiology, molecular drivers, and targeted therapy. Cancers 2021, 13, 1931. [Google Scholar]
- Kempf, W.; Torricelli, R.; Zettl, A.; Zimmermann, A.-K.; Berisha, A.; Ghielmini, M. Primary Cutaneous Anaplastic Lymphoma Kinase–Positive Large B-Cell Lymphoma. Am. J. Dermatopathol. 2019, 41, 602–605. [Google Scholar] [PubMed]
- Wang, L.; Xiao, H.; Zhang, X.; Wang, C.; Huang, H. The role of telomeres and telomerase in hematologic malignancies and hematopoietic stem cell transplantation. J. Hematol. Oncol. 2014, 7, 61. [Google Scholar]
- Ropio, J.; Merlio, J.-P.; Soares, P.; Chevret, E. Telomerase activation in hematological malignancies. Genes 2016, 7, 61. [Google Scholar]
- Chevret, E.; Andrique, L.; Prochazkova-Carlotti, M.; Ferrer, J.; Cappellen, D.; Laharanne, E.; Idrissi, Y.; Boettiger, A.; Sahraoui, W.; Ruiz, F.; et al. Telomerase functions beyond telomere maintenance in primary cutaneous T-cell lymphoma. Blood J. Am. Soc. Hematol. 2014, 123, 1850–1859. [Google Scholar] [CrossRef] [PubMed]
- Poglio, S.; Prochazkova-Carlotti, M.; Cherrier, F.; Gros, A.; Laharanne, E.; Pham-Ledard, A.; Beylot-Barry, M.; Merlio, J.-P. Xenograft and cell culture models of Sézary syndrome reveal cell of origin diversity and subclonal heterogeneity. Leukemia 2021, 35, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- Ropio, J.; Chebly, A.; Ferrer, J.; Prochazkova-Carlotti, M.; Idrissi, Y.; Azzi-Martin, L.; Cappellen, D.; Pham-Ledard, A.; Soares, P.; Merlio, J.P.; et al. Reliable blood cancer cells’ telomere length evaluation by qPCR. Cancer Med. 2020, 9, 3153–3162. [Google Scholar] [CrossRef] [Green Version]
- Chebly, A.; Ropio, J.; Peloponese, J.M.; Poglio, S.; Prochazkova-Carlotti, M.; Cherrier, F.; Ferrer, J.; Idrissi, Y.; Segal-Bendirdjian, E.; Chouery, E.; et al. Exploring hTERT promoter methylation in cutaneous T-cell lymphomas. Mol. Oncol. 2021, 16, 1931–1946. [Google Scholar]
- Chebly, A.; Prochazkova-Carlotti, M.; Idrissi, Y.; Bresson-Bepoldin, L.; Poglio, S.; Farra, C.; Beylot-Barry, M.; Merlio, J.-P.; Tomb, R.; Chevret, E. Targeting Epigenetic Modifiers Can Reduce the Clonogenic Capacities of Sézary Cells. Front. Oncol. 2021, 11, 775253. [Google Scholar]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Penna, G.; Gerace, D.; Allegra, A.G.; Musolino, C. Telomerase and telomere biology in hematological diseases: A new therapeutic target. Leuk. Res. 2017, 56, 60–74. [Google Scholar]
- Mocellin, S.; Verdi, D.; Pooley, K.A.; Landi, M.T.; Egan, K.M.; Baird, D.M.; Prescott, J.; De Vivo, I.; Nitti, D. Telomerase reverse transcriptase locus polymorphisms and cancer risk: A field synopsis and meta-analysis. J. Natl. Cancer Inst. 2012, 104, 840–854. [Google Scholar]
- Heidenreich, B.; Kumar, R. TERT promoter mutations in telomere biology. Mutat. Res./Rev. Mutat. Res. 2017, 771, 15–31. [Google Scholar]
- Mosrati, M.A.; Malmström, A.; Lysiak, M.; Krysztofiak, A.; Hallbeck, M.; Milos, P.; Hallbeck, A.-L.; Bratthäll, C.; Strandéus, M.; Stenmark-Askmalm, M.; et al. TERT promoter mutations and polymorphisms as prognostic factors in primary glioblastoma. Oncotarget 2015, 6, 16663. [Google Scholar]
- Yan, S.; Han, B.; Wu, Y.; Zhou, D.; Zhao, Y. Telomerase gene mutation screening and telomere overhang detection in Chinese patients with acute myeloid leukemia. Leuk. Lymphoma 2013, 54, 1437–1441. [Google Scholar] [CrossRef]
- Liu, H.; Liu, Q.; Ge, Y.; Zhao, Q.; Zheng, X.; Zhao, Y. hTERT promotes cell adhesion and migration independent of telomerase activity. Sci. Rep. 2016, 6, 22886. [Google Scholar] [CrossRef]
- Chevret, E.; Merlio, J.-P. Sézary syndrome: Translating genetic diversity into personalized medicine. J. Investig. Dermatol. 2016, 136, 1319–1324. [Google Scholar] [CrossRef]
- Herrera, A.; Cheng, A.; Mimitou, E.P.; Seffens, A.; George, D.; Bar-Natan, M.; Heguy, A.; Ruggles, K.V.; Scher, J.U.; Hymes, K.; et al. Multimodal single-cell analysis of cutaneous T-cell lymphoma reveals distinct subclonal tissue-dependent signatures. Blood J. Am. Soc. Hematol. 2021, 138, 1456–1464. [Google Scholar]
- Najidh, S.; Tensen, C.P.; van der Sluijs-Gelling, A.J.; Teodosio, C.; Cats, D.; Mei, H.; Kuipers, T.B.; Out-Luijting, J.J.; Zoutman, W.H.; van Hall, T.; et al. Improved Sézary cell detection and novel insights into immunophenotypic and molecular heterogeneity in Sézary syndrome. Blood J. Am. Soc. Hematol. 2021, 138, 2539–2554. [Google Scholar]
- Subasri, M.; Shooshtari, P.; Watson, A.J.; Betts, D.H. Analysis of TERT Isoforms across TCGA, GTEx and CCLE Datasets. Cancers 2021, 13, 1853. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, S.; Kumar, M.S.; Peters, G.; Mayur, Y. Targeting telomerase for its advent in cancer therapeutics. Med. Res. Rev. 2020, 40, 1871–1919. [Google Scholar] [PubMed]
- Gomez, D.; Lemarteleur, T.; Lacroix, L.; Mailliet, P.; Mergny, J.L.; Riou, J.F. Telomerase downregulation induced by the G-quadruplex ligand 12459 in A549 cells is mediated by hTERT RNA alternative splicing. Nucleic Acids Res. 2004, 32, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Shen, J.; Cao, J.; Zhou, G.; Lei, T.; Sun, Y.; Gao, H.; Ding, Y.; Xu, W.; Zhan, Z. Alternative splicing of human telomerase reverse transcriptase in gliomas and its modulation mediated by CX-5461. J. Exp. Clin. Cancer Res. 2018, 37, 78. [Google Scholar] [CrossRef] [Green Version]
- Willemze, R.; Hodak, E.; Zinzani, P.; Specht, L.; Ladetto, M. Primary cutaneous lymphomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv30–iv40. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus/Genotype | Cell Lines n (%) | Controls n (%) | Patients n (%) | OR (95% CI) | p-Value |
|---|---|---|---|---|---|
| rs2735940 | n = 8 | n = 101 | n = 66 | ||
| TT | 2 (25) | 40 (39.6) | 11 (16.7) | 1.00 a | |
| TC | 6 (75) | 48 (47.5) | 39 (59.1) | 3.00 (1.31–6.89) | 0.010 |
| CC | 0 | 13 (12.9) | 16 (24.2) | 3.79 (1.35–10.6) | 0.011 |
| Dominant model (C carrier vs. TT b) | 61 (60.4)/11 (39.6) | 55 (83.3)/11 (16.7) | 3.20 (1.44–7.08) | 0.004 | |
| rs2853669 | n = 8 | n = 96 | n = 66 | ||
| TT | 0 | 40 (41.7) | 28 (42.4) | 1.00 a | |
| TC | 8 (100) | 42 (43.7) | 29 (43.9) | 0.84 (0.41–1.75) | 0.649 |
| CC | 0 | 14 (14.6) | 9 (13.6) | 0.80 (0.29–2.21) | 0.660 |
| Dominant model (C carrier vs. TT b) | 56 (58.3)/40 (41.7) | 38 (57.6)/28 (42.4) | 0.83 (0.42–1.64) | 0.595 | |
| rs2853672 | n = 8 | n = 101 | n = 66 | ||
| GG | 2 (25) | 38 (37.6) | 16 (24.2) | 1.00 a | |
| GT | 6 (75) | 50 (49.5) | 38 (57.6) | 2.04 (0.94–4.44) | 0.069 |
| TT | 0 | 13 (12.9) | 12 (18.2) | 2.67 (0.94–7.62) | 0.063 |
| Dominant model (T carrier vs. GG b) | 63 (62.4)/38 (37.6) | 50 (75.8)/16 (24.2) | 2.18 (1.04–4.58) | 0.039 | |
| rs2853676 | n = 8 | n = 101 | n = 66 | ||
| GG | 4 (50) | 47 (46.5) | 40 (40.6) | 1.00 a | |
| AG | 3 (37.5) | 42 (41.6) | 26 (39.4) | 0.59 (0.29–1.19) | 0.141 |
| AA | 1 (12.5) | 12 (11.9) | 0 | 0.00 | 0.999 |
| Dominant model (A carrier vs. GG b) | 54 (53.5)/47 (46.5) | 26 (39.4)/40 (60.6) | 0.46 (0.23–0.92) | 0.028 | |
| rs10069690 | n = 8 | n = 101 | n = 66 | ||
| CC | 0 | 50 (49.5) | 36 (54.5) | 1.00 a | |
| CT | 8 (100) | 38 (37.6) | 29 (43.9) | 1.28 (0.64–2.57) | 0.479 |
| TT | 0 | 13 (12.9) | 1 (1.5) | 0.14 (0.017–1.21) | 0.074 |
| Dominant model (T carrier vs. CC b) | 51 (50.5)/50 (49.5) | 30 (45.5)/36 (54.5) | 1.02 (0.52–1.98) | 0.963 |
| Mutation Rate (%) | Mutation | |
|---|---|---|
| Patients | 1/67 (1.5%) | |
| LPDs | 0/18 (0%) | - |
| cALCL | 0/10 (0%) | - |
| LyP | 0/8 (0%) | - |
| MF | 0/32 (0%) | - |
| MF | 0/24 (0%) | - |
| T-MF | 0/8 (0%) | - |
| SS | 1/17 (5.9%) | −146 C > T |
| Cell lines | 1/8 (12.5%) | |
| cALCL | 0/4 (0%) | - |
| T-MF | 1/1 (100%) | −146 C > T |
| SS | 0/3 (0%) | - |
| Cells | Total hTERT Transcripts (Sum of α+β+, α−β+, and α+β−) | α+β+ | α−β+ | α+β− |
|---|---|---|---|---|
| % (p-Value) | % (p Value) | |||
| Mac1 | 100 | 22.8 | 8.0 | 69.2 |
| Mac2A | 100 (0.0022) | 63.2 (0.5887) | 5.9 (0.0022) | 30.9 (0.0022) |
| Mac2B | 100 (0.0260) | 70.5 (0.1667) | 12.0 (0.0628) | 17.5 (0.0022) |
| Healthy controls | 100 | 2.9 | 0.0 | 97.1 |
| SS patients | 100 (0.0120) | 35.0 (0.2507) | 4.9 (0.0050) | 60.1 (0.0290) |
| Cells | Total hTERT Transcripts (Sum of α+β+, α−β+ and α+β−) | α+β+ | α−β+ | α+β− |
|---|---|---|---|---|
| % (p Value) | % (p Value) | |||
| shControl | 50.9 | 9.9 | 39.2 | |
| sh1 | 100 (0.1797) | 48.3 (0.4857) | 17.7 (0.0237) | 34 (0.2196) |
| sh2 | 100 (0.1797) | 54.5 (0.9448) | 5.5 (0.0003) | 40 (0.7255) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ropio, J.; Prochazkova-Carlotti, M.; Batista, R.; Pestana, A.; Chebly, A.; Ferrer, J.; Idrissi, Y.; Cappellen, D.; Durães, C.; Boaventura, P.; et al. Spotlight on hTERT Complex Regulation in Cutaneous T-Cell Lymphomas. Genes 2023, 14, 439. https://doi.org/10.3390/genes14020439
Ropio J, Prochazkova-Carlotti M, Batista R, Pestana A, Chebly A, Ferrer J, Idrissi Y, Cappellen D, Durães C, Boaventura P, et al. Spotlight on hTERT Complex Regulation in Cutaneous T-Cell Lymphomas. Genes. 2023; 14(2):439. https://doi.org/10.3390/genes14020439
Chicago/Turabian StyleRopio, Joana, Martina Prochazkova-Carlotti, Rui Batista, Ana Pestana, Alain Chebly, Jacky Ferrer, Yamina Idrissi, David Cappellen, Cecília Durães, Paula Boaventura, and et al. 2023. "Spotlight on hTERT Complex Regulation in Cutaneous T-Cell Lymphomas" Genes 14, no. 2: 439. https://doi.org/10.3390/genes14020439