
Effects of Elexacaftor/Tezacaftor/Ivacaftor on Cardiorespiratory Polygraphy Parameters and Respiratory Muscle Strength in Cystic Fibrosis Patients with Severe Lung Disease

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Design and Participants
2.2. Inclusion Criteria
- Diagnosis of CF based on clinical presentation of CF (positive newborn screening test, suggestive signs or symptoms, and family history) plus positive sweat chloride test (Cl ≥ 60 mmol/L) or clinical presentation plus borderline sweat chloride results (Cl > 30 mmol/L and Cl < 60 mmol/L) plus two CF-causing CFTR mutations or CFTR genotype undefined or unknown based on genetic analysis plus positive CFTR physiologic testing showing CFTR dysfunction [38];
- The inclusion criteria for compassionate use included a genotype heterozygous for F508del and one qualifying MF allele after whole CFTR gene scanning, age > 12 years, and severe lung disease, defined as either highest percentage predicted FEV1 (ppFEV1) < 40% in the preceding 2 months or being on a lung transplant waiting list [19].
2.3. Dosing
2.4. Outcome Measures
2.5. Nocturnal Cardiorespiratory Polygraphy
2.6. MIP and MEP
2.7. ppFEV1
2.8. 6MWT
2.9. Statistical Analysis
3. Results
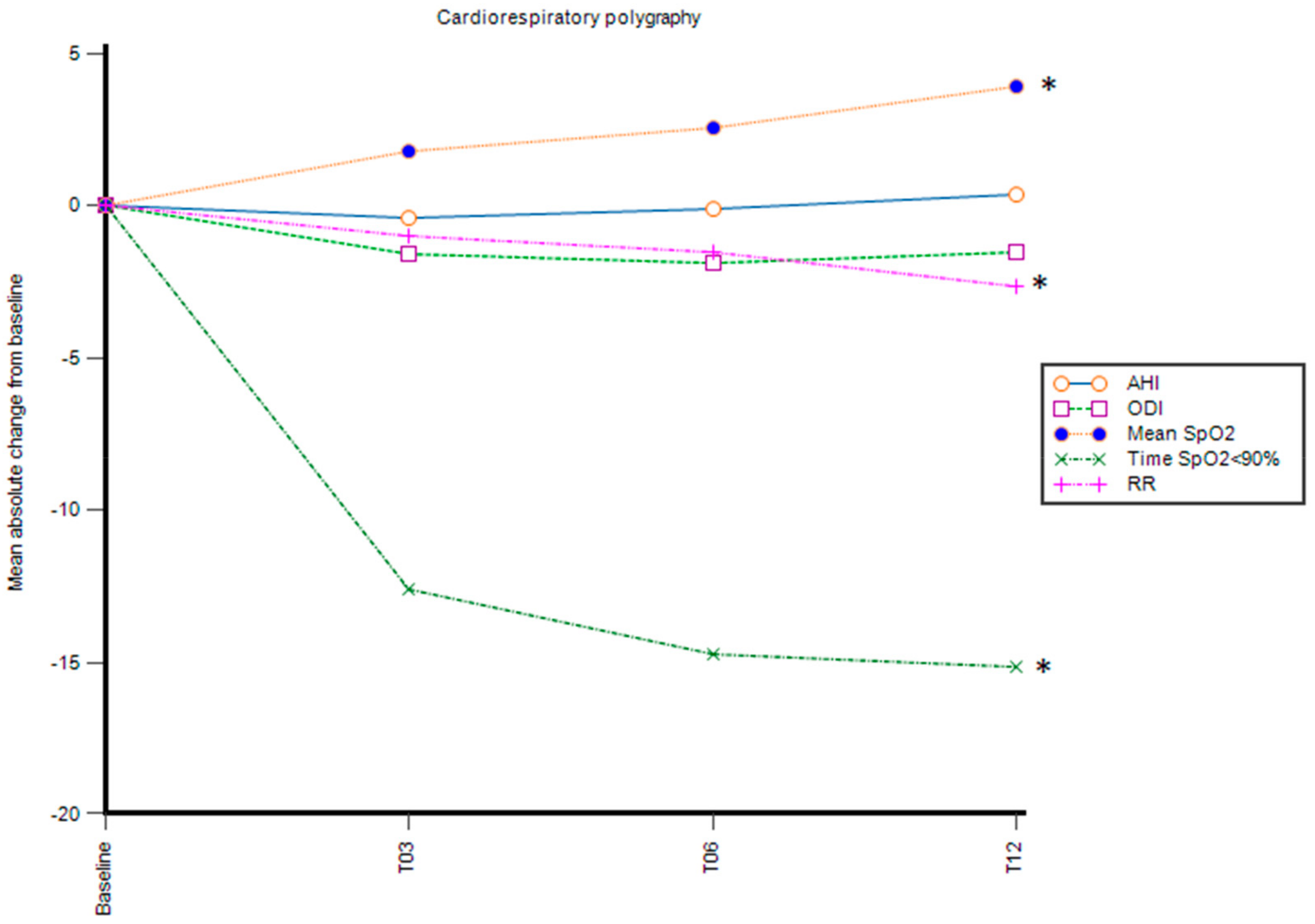
3.1. Nocturnal Cardiorespiratory Polygraphy Parameters
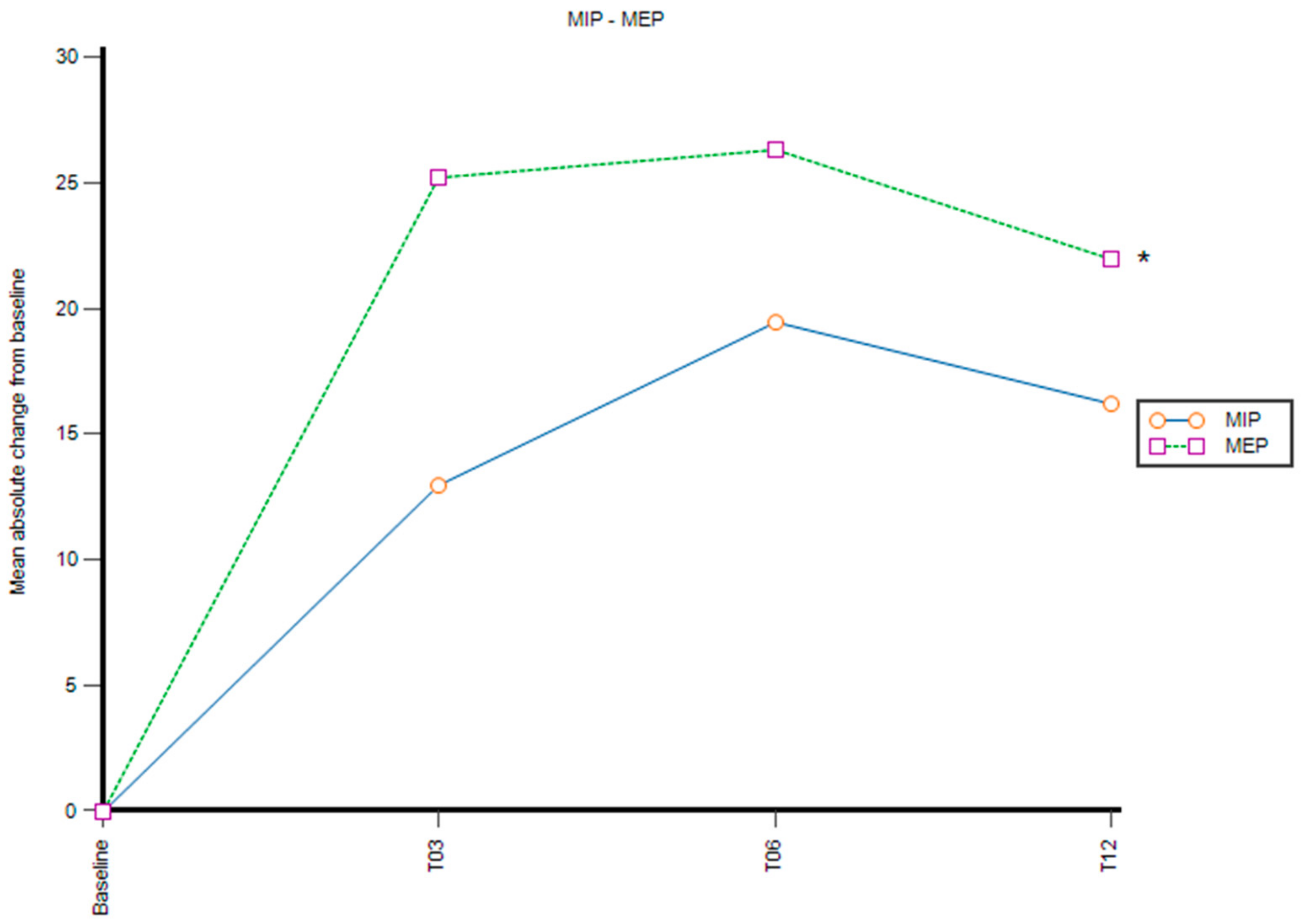
3.2. MIP and MEP
3.3. 6MWT
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L. Identification of the cystic fibrosis gene: Cloning and characterization of complementary. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Mutation Database: Statistics. Available online: http://genet.sickkids.on.ca/StatisticsPage.html (accessed on 28 November 2022).
- CFTR2 Variant List History|CFTR2. Available online: http://www.cftr2.org/ (accessed on 28 November 2022).
- Marson, F.A.L.; Bertuzzo, C.S.; Ribeiro, J.D. Classification of CFTR mutation classes. Lancet Respir. Med. 2016, 4, e37–e38. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Zolin, A.; Cuppens, H.; Olesen, H.V.; Viviani, L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J. Cyst. Fibros. 2014, 13, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Zolin, A.; Orenti, A.; Naehrlich, L.; Jung, A.; van Rens, J.; Fox, A.; Krasnyk, M.; Cosgriff, R.; Hatziagorou, E.; Mei-Zahav, M.; et al. ECFSPR Annual Report 2018. Available online: https://www.ecfs.eu/projects/ecfspatient-registry/annual-reports (accessed on 19 October 2022).
- Scotet, V.; L’Hostis, C.; Férec, C. The Changing Epidemiology of Cystic Fibrosis: Incidence, Survival and Impact of the CFTR Gene Discovery. Genes 2020, 11, 589. [Google Scholar] [CrossRef] [PubMed]
- Annual Data Report 2019. Available online: https://cff.org/Research/Researcher-Resources/PatientRegistry/2019-Patient-Registry-Annual-Data-Report.pdf (accessed on 28 November 2022).
- 2018 Registry Annual Data Report. Available online: https://www.cysticfibrosis.org.uk/sites/default/files/2020-12/2018%20Registry%20Annual%20Data%20Report.pdf (accessed on 28 November 2022).
- Castellani, C.; Duff, A.J.A.; Bell, S.C.; Heijerman, H.G.M.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS best practice guidelines: The 2018 revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef]
- Sutharsan, S.; McKone, E.F.; Downey, D.G.; Duckers, J.; MacGregor, G.; Tullis, E.; Van Braeckel, E.; Wainwright, C.E.; Watson, D.; Ahluwalia, N.; et al. Efficacy and safety of elexacaftor plus tezacaftor plus ivacaftor versus tezacaftor plus ivacaftor in people with cystic fibrosis homozygous for F508del-CFTR: A 24-week, multicentre, randomised, double-blind, active-controlled, phase 3b trial. Lancet Respir. Med. 2022, 10, 267–277. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Carnovale, V.; Iacotucci, P.; Terlizzi, V.; Colangelo, C.; Ferrillo, L.; Pepe, A.; Francalanci, M.; Taccetti, G.; Buonaurio, S.; Celardo, A.; et al. Elexacaftor/Tezacaftor/Ivacaftor in Patients with Cystic Fibrosis Homozygous for the F508del Mutation and Advanced Lung Disease: A 48-Week Observational Study. J. Clin. Med. 2022, 11, 1021. [Google Scholar] [CrossRef] [PubMed]
- Carnovale, V.; Iacotucci, P.; Terlizzi, V.; Colangelo, C.; Medio, P.; Ferrillo, L.; De Gregorio, F.; Francalanci, M.; Taccetti, G.; Buonaurio, S.; et al. Effectiveness and safety of elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease with the Phe508del/minimal function genotype. Respir. Med. 2021, 189, 106646. [Google Scholar] [CrossRef]
- Terlizzi, V.; Colangelo, C.; Marsicovetere, G.; D’Andria, M.; Francalanci, M.; Innocenti, D.; Masi, E.; Avarello, A.; Taccetti, G.; Amato, F.; et al. Effectiveness of Elexacaftor/Tezacaftor/Ivacaftor Therapy in Three Subjects with the Cystic Fibrosis Genotype Phe508del/Unknown and Advanced Lung Disease. Genes 2021, 12, 1178. [Google Scholar] [CrossRef]
- Comegna, M.; Terlizzi, V.; Salvatore, D.; Colangelo, C.; Di Lullo, A.M.; Zollo, I.; Taccetti, G.; Castaldo, G.; Amato, F. Elexacaftor-Tezacaftor-Ivacaftor Therapy for Cystic Fibrosis Patients with The F508del/Unknown Genotype. Antibiotics 2021, 10, 828. [Google Scholar] [CrossRef]
- Terlizzi, V.; Amato, F.; Castellani, C.; Ferrari, B.; Galietta, L.J.V.; Castaldo, G.; Taccetti, G. Ex vivo model predicted in vivo efficacy of CFTR modulator therapy in a child with rare genotype. Mol. Genet. Genom. Med. 2021, 9, e1656. [Google Scholar] [CrossRef]
- European Medicines Agency. Kaftrio. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/kaftrio (accessed on 28 November 2022).
- Welsner, M.; Schulte, T.; Dietz-Terjung, S.; Weinreich, G.; Stehling, F.; Taube, C.; Strassburg, S.; Schoebel, C.; Sutharsan, S. Effect of Triple Combination CFTR Modulator Therapy on Sleep in Adult Patients with Cystic Fibrosis. Respiration 2022, 101, 766–774. [Google Scholar] [CrossRef]
- Causer, A.J.; Shute, J.K.; Cummings, M.H.; Shepherd, A.I.; Wallbanks, S.R.; Pulsford, R.M.; Bright, V.; Connett, G.; Saynor, Z.L. Elexacaftor-Tezacaftor-Ivacaftor improves exercise capacity in adolescents with cystic fibrosis. Pediatr. Pulmonol. 2022, 57, 2652–2658. [Google Scholar] [CrossRef]
- Cohen-Cymberknoh, M.; Atia, O.; Gileles-Hillel, A.; Kerem, E.; Reiter, J. Sleep disorders in patients with primary ciliary dyskinesia, cystic fibrosis with and without pancreatic insufficiency. Respir. Med. 2019, 151, 96–101. [Google Scholar] [CrossRef]
- Reiter, J.; Breuer, O.; Cohen-Cymberknoh, M.; Forno, E.; Gileles-Hillel, A. Sleep in children with cystic fibrosis: More under the covers. Pediatr. Pulmonol. 2022, 57, 1944–1951. [Google Scholar] [CrossRef] [PubMed]
- Garde, A.J.B.; Gibson, N.A.; Samuels, M.P.; Evans, H.J. Recent advances in paediatric sleep disordered breathing. Breathe 2022, 18, 220151. [Google Scholar] [CrossRef] [PubMed]
- Spicuzza, L.; Sciuto, C.; Leonardi, S.; La Rosa, M. Early Occurrence of Obstructive Sleep Apnea in Infants and Children With Cystic Fibrosis. Arch. Pediatr. Adolesc. Med. 2012, 166, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Lands, L.; Desmond, K.J.; Demizio, D.; Pavilanis, A.; Coates, A.L. The effects of nutritional status and hyperinflation on respiratory muscle strength in children and young adults. Am. Rev. Respir. Dis. 1990, 141, 1506–1509. [Google Scholar] [CrossRef] [PubMed]
- Pinet, C.; Cassart, M.; Scillia, P.; Lamotte, M.; Knoop, C.; Casimir, G.; Mélot, C.; Estenne, M. Function and bulk of respiratory and limb muscles in patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 989–994. [Google Scholar] [CrossRef] [PubMed]
- Gusev, E.; Liang, F.; Bhattarai, S.; Broering, F.E.; Leduc-Gaudet, J.P.; Hussain, S.N.; Radzioch, D.; Petrof, B.J. Characterization of skeletal muscle wasting pathways in diaphragm and limb muscles of cystic fibrosis mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2022, 322, R551–R561. [Google Scholar] [CrossRef]
- Dunnink, M.A.; Doeleman, W.R.; Trappenburg, J.C.; de Vries, W.R. Respiratory muscle strength in stable adolescent and adult patients with cystic fibrosis. J. Cyst. Fibros. 2009, 8, 31–36. [Google Scholar] [CrossRef]
- Dekerlegand, R.L.; Hadjiliadis, D.; Swisher, A.K.; Parrott, J.S.; Heuer, A.J.; Myslinski, M.J. Inspiratory muscle strength relative to disease severity in adults with stable cystic fibrosis. J. Cyst. Fibros. 2015, 14, 639–645. [Google Scholar] [CrossRef]
- Sovtic, A.; Minic, P.; Markovic-Sovtic, G.; Trajkovic, G.Z. Respiratory Muscle Strength and Exercise Performance in Cystic Fibrosis-A Cross Sectional Study. Front. Pediatr. 2018, 6, 244. [Google Scholar] [CrossRef]
- Andrade Lima, C.; Dornelas de Andrade, A.; Campos, S.L.; Brandão, D.C.; Mourato, I.P.; Britto, M.C.A. Six-minute walk test as a determinant of the functional capacity of children and adolescents with cystic fibrosis: A systematic review. Respir. Med. 2018, 137, 83–88. [Google Scholar] [CrossRef]
- Martin, C.; Chapron, J.; Hubert, D.; Kanaan, R.; Honoré, I.; Paillasseur, J.L.; Aubourg, F.; Dinh-Xuan, A.T.; Dusser, D.; Fajac, I.; et al. Prognostic value of six minute walk test in cystic fibrosis adults. Respir. Med. 2013, 107, 1881–1887. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181S, S4–S15.e1. [Google Scholar] [CrossRef] [PubMed]
- Berry, R.B.; Budhiraja, R.; Gottlieb, D.J.; Gozal, D.; Iber, C.; Kapur, V.K.; Marcus, C.L.; Mehra, R.; Parthasarathy, S.; Quan, S.F.; et al. Rules for scoring respiratory events in sleep: Update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events: Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J. Clin. Sleep Med. 2012, 8, 597–619. [Google Scholar] [CrossRef] [PubMed]
- American Thoracic Society/European Respiratory Society. ATS/ERS Statement on respiratory muscle testing. Am. J. Respir. Crit. Care Med. 2002, 166, 518–624. [Google Scholar] [CrossRef]
- Miller, M.R.; Hankinson, J.; Brusasco, V.; Burgos, F.; Casaburi, R.; Coates, A.; Crapo, R.; Enright, P.; van der Grinten, C.P.; Gustafsson, P.; et al. Standardisation of spirometry. Eur. Respir. J. 2005, 26, 319–338. [Google Scholar] [CrossRef]
- ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories. ATS statement: Guidelines for the six-minute walk test [published correction appears in Am J Respir Crit Care Med. 2016 May 15;193(10):1185]. Am. J. Respir. Crit. Care Med. 2002, 166, 111–117. [Google Scholar] [CrossRef]
- Cronly, J.A.; Duff, A.J.; Riekert, K.A.; Fitzgerald, A.P.; Perry, I.J.; Lehane, E.A.; Horgan, A.; Howe, B.A.; Ni Chroinin, M.; Savage, E. Health-Related Quality of Life in Adolescents and Adults With Cystic Fibrosis: Physical and Mental Health Predictors. Respir. Care 2019, 64, 406–415. [Google Scholar] [CrossRef]
- Dancey, D.R.; Tullis, E.D.; Heslegrave, R.; Thornley, K.; Hanly, P.J. Sleep quality and daytime function in adults with cystic fibrosis and severe lung disease. Eur. Respir. J. 2002, 19, 504–510. [Google Scholar] [CrossRef]
- Fraser, K.L.; Tullis, D.E.; Sasson, Z.; Hyland, R.H.; Thornley, K.S.; Hanly, P.J. Pulmonary hypertension and cardiac function in adult cystic fibrosis: Role of hypoxemia. Chest 1999, 115, 1321–1328. [Google Scholar] [CrossRef]
- Young, A.C.; Wilson, J.W.; Kotsimbos, T.C.; Naughton, M.T. The impact of nocturnal oxygen desaturation on quality of life in cystic fibrosis. J. Cyst. Fibros. 2011, 10, 100–106. [Google Scholar] [CrossRef]
- Vandeleur, M.; Walter, L.M.; Armstrong, D.S.; Robinson, P.; Nixon, G.M.; Horne, R.S.C. What keeps children with cystic fibrosis awake at night? J. Cyst. Fibros. 2017, 16, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Urquhart, D.S.; Montgomery, H.; Jaffé, A. Assessment of hypoxia in children with cystic fibrosis. Arch. Dis. Child. 2005, 90, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Sateia, M.J. International classification of sleep disorders-third edition: Highlights and modifications. Chest 2014, 146, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Ramos, R.T.; Santana, M.A.; Almeida Pde, C.; Machado Ade, S., Jr.; Araújo-Filho, J.B.; Salles, C. Nocturnal hypoxemia in children and adolescents with cystic fibrosis. J. Bras. De Pneumol. 2013, 39, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.; Gileles-Hillel, A.; Cohen-Cymberknoh, M.; Rosen, D.; Kerem, E.; Gozal, D.; Forno, E. Sleep disorders in cystic fibrosis: A systematic review and meta-analysis. Sleep Med. Rev. 2020, 51, 101279. [Google Scholar] [CrossRef] [PubMed]
- Bradley, S.; Solin, P.; Wilson, J.; Johns, D.; Walters, E.H.; Naughton, M.T. Hypoxemia and hypercapnia during exercise and sleep in patients with cystic fibrosis. Chest 1999, 116, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Uyan, Z.S.; Ozdemir, N.; Ersu, R.; Akpinar, I.; Keskin, S.; Cakir, E.; Karadağ, B.; Karakoç, F.; Dağli, E. Factors that correlate with sleep oxygenation in children with cystic fibrosis. Pediatr. Pulmonol. 2007, 42, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Shakkottai, A.; Nasr, S.Z.; Hassan, F.; Irani, S.; O’Brien, L.M.; Chervin, R.D. Sleep-disordered breathing in cystic fibrosis. Sleep Med. 2020, 74, 57–65. [Google Scholar] [CrossRef]
- Papale, M.; Parisi, G.F.; Spicuzza, L.; Licari, A.; Bongiovanni, A.; Mulè, E.; Rotolo, N.; Manti, S.; Leonardi, S. Lung clearance index evaluation in detecting nocturnal hypoxemia in cystic fibrosis patients: Toward a new diagnostic tool. Respir. Med. 2020, 164, 105906. [Google Scholar] [CrossRef]
- van der Giessen, L.; Bakker, M.; Joosten, K.; Hop, W.; Tiddens, H. Nocturnal oxygen saturation in children with stable cystic fibrosis. Pediatr. Pulmonol. 2012, 47, 1123–1130. [Google Scholar] [CrossRef]
- Fauroux, B.; Pepin, J.L.; Boelle, P.Y.; Cracowski, C.; Murris-Espin, M.; Nove-Josserand, R.; Stremler, N.; Simon, T.; Burgel, P.R. Sleep quality and nocturnal hypoxaemia and hypercapnia in children and young adults with cystic fibrosis. Arch. Dis. Child. 2012, 97, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, R.R.B.; Liberato, F.M.G.; de Freitas Coelho, P.; Vidal, P.D.R.; de Carvalho, R.B.C.O.; Donadio, M.V.F. Sleep-disordered breathing and markers of morbidity in children and adolescents with cystic fibrosis. Pediatr. Pulmonol. 2020, 55, 1974–1983. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Descalço, A.; Salgueiro, M.; Pereira, L.; Barreto, C.; Bandeira, T.; Ferreira, R. Respiratory sleep disturbance in children and adolescents with cystic fibrosis. Rev. Port. De Pneumol. 2016, 22, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.R.; Durieu, I.; Chiron, R.; Ramel, S.; Danner-Boucher, I.; Prevotat, A.; Grenet, D.; Marguet, C.; Reynaud-Gaubert, M.; Macey, J.; et al. Rapid Improvement after Starting Elexacaftor-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and Advanced Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 64–73. [Google Scholar] [CrossRef]
- Shteinberg, M.; Taylor-Cousar, J.L. Impact of CFTR modulator use on outcomes in people with severe cystic fibrosis lung disease. Eur. Respir. Rev. 2020, 29, 190112. [Google Scholar] [CrossRef]
- Stylemans, D.; Darquenne, C.; Schuermans, D.; Verbanck, S.; Vanderhelst, E. Peripheral lung effect of elexacaftor/tezacaftor/ivacaftor in adult cystic fibrosis. J. Cyst. Fibros. 2022, 21, 160–163. [Google Scholar] [CrossRef]
- Montgomery, S.T.; Mall, M.A.; Kicic, A.; Stick, S.M. Hypoxia and sterile inflammation in cystic fibrosis airways: Mechanisms and potential therapies. Eur. Respir. J. 2017, 49, 1600903. [Google Scholar] [CrossRef]
- Moliteo, E.; Sciacca, M.; Palmeri, A.; Papale, M.; Manti, S.; Parisi, G.F.; Leonardi, S. Cystic Fibrosis and Oxidative Stress: The Role of CFTR. Molecules 2022, 27, 5324. [Google Scholar] [CrossRef]
- Ziegler, B.; Perin, C.; Casarotto, F.C.; Fagondes, S.C.; Menna-Barreto, S.S.; Dalcin, P. Pulmonary hypertension as estimated by Doppler echocardiography in adolescent and adult patients with cystic fibrosis and their relationship with clinical, lung function and sleep findings. Clin. Respir. J. 2018, 12, 754–761. [Google Scholar] [CrossRef]
- Jeffery, T.K.; Wanstall, J.C. Pulmonary vascular remodeling: A target for therapeutic intervention in pulmonary hypertension. Pharmacol. Ther. 2001, 92, 1–20. [Google Scholar] [CrossRef]
- Wark, P.A.B.; Cookson, K.; Thiruchelvam, T.; Brannan, J.; Dorahy, D.J. Lumacaftor/ Ivacaftor improves exercise tolerance in patients with Cystic Fibrosis and severe airflow obstruction. BMC Pulm. Med. 2019, 19, 106. [Google Scholar] [CrossRef] [PubMed]
- Gur, M.; Masarweh, K.; Toukan, Y.; Nir, V.; Bar-Yoseph, R.; Hanna, M.; Manor, E.; Hakim, F.; Bentur, L. Six-minute walk, lung clearance index, and QOL in bronchiolitis obliterans and cystic fibrosis. Pediatr. Pulmonol. 2019, 54, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Gambazza, S.; Turrin, V.; Speziali, C.; Brivio, A.; Valmarana, L.; Carta, F.; Bulfamante, A.M.C.; Colombo, C. Expiratory muscle strength and functional exercise tolerance in adults with cystic fibrosis: A cross-sectional study. Physiother. Res. Int. 2018, 23, e1720. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef] [PubMed]
- Stanojevic, S.; Vukovojac, K.; Sykes, J.; Ratjen, F.; Tullis, E.; Stephenson, A.L. Projecting the impact of delayed access to elexacaftor/tezacaftor/ivacaftor for people with Cystic Fibrosis. J. Cyst. Fibros. 2021, 20, 243–249. [Google Scholar] [CrossRef]
- Giordano, S.; Amato, F.; Elce, A.; Monti, M.; Iannone, C.; Pucci, P.; Seia, M.; Angioni, A.; Zarrilli, F.; Castaldo, G.; et al. Molecular and functional analysis of the large 5′ promoter region of CFTR gene revealed pathogenic mutations in CF and CFTR-related disorders. J. Mol. Diagn. 2013, 15, 331–340. [Google Scholar] [CrossRef]
- Parisi, G.F.; Mòllica, F.; Giallongo, A.; Papale, M.; Manti, S.; Leonardi, S. Cystic fibrosis transmembrane conductance regulator (CFTR): Beyond cystic fibrosis. Egypt. J. Med. Hum. Genet. 2022, 23, 94. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Demographics | |
|---|---|
| Patients | 9 |
| Males (%) | 2 (22.2) |
| Age at start of treatment, mean (SD), yrs | 30.3 (6.5) |
| Age < 18, yrs (%) | 0 (0) |
| ppFEV1, mean (SD) | 34.6 (5.1) |
| ppFEV1 < 40% (%) | 9 (100) |
| P. aeruginosa colonization (%) | 4 (44.4) |
| CFQ-R, mean (SD) | 73.3 (17.9) |
| BMI, mean (SD), kg/m2 | 19.9 (2.5) |
| AHI, mean (SD) | 1.4 (1.8) |
| ODI, mean (SD) | 5.5 (5.8) |
| Mean nocturnal SpO2%, mean (SD) | 92.4 (2.9) |
| time SpO2 ≤ 90%, min (SD) | 15.7 (28.1) |
| RR, mean (SD) | 26 (1.9) |
| Ventilation (%) | 7 (77.8) |
| 6MWT, mean (SD), m | 483 (67.5) |
| MIP, mean (SD), mmHg | 72.7 (32.1) |
| MEP, mean (SD), mmHg | 58 (27.7) |
| Outcomes | Baseline (SD) | Month 3 | Month 6 | Month 12 | Change from Baseline | p Value |
|---|---|---|---|---|---|---|
| AHI | 1.4 (1.8) | 1 (0.8) | 1.3 (0.8) | 1.7 (0.8) | 0.3 | p > 0.05 |
| ODI | 5.5 (5.8) | 3.9 (1.3) | 3.8 (1.7) | 4 (2.9) | −1.5 | p > 0.05 |
| Mean nocturnal SpO2 | 92.4 (2.9) | 94.2 (1.5) | 94.8 (2) | 96.4 (1.6) | 4 | p = 0.03 * |
| time SpO2 ≤ 90%, min | 15.7 (28.1) | 3.1 (2.9) | 1.1 (1.1) | 0.5 (0.8) | −15.2 | p = 0.03 * |
| RR | 26 (1.9) | 25 (2.1) | 24.4 (1.4) | 23.3 (0.7) | −2.7 | p = 0.004 * |
| MIP, mmHg | 72.7 (32.1) | 85.7 (34.8) | 92.1 (30.3) | 88.8 (27.6) | 16.1 | p > 0.05 |
| MEP, mmHg | 58 (27.7) | 83.2 (21.3) | 84.3 (32.5) | 80 (35.8) | 22 | p = 0.001 * |
| 6MWT, m | 483 (67.5) | 513.3 (46.1) | 516.7 (32.8) | 573 (60.8) | 90 | p = 0.008 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giallongo, A.; Parisi, G.F.; Papale, M.; Manti, S.; Mulé, E.; Aloisio, D.; Terlizzi, V.; Rotolo, N.; Leonardi, S. Effects of Elexacaftor/Tezacaftor/Ivacaftor on Cardiorespiratory Polygraphy Parameters and Respiratory Muscle Strength in Cystic Fibrosis Patients with Severe Lung Disease. Genes 2023, 14, 449. https://doi.org/10.3390/genes14020449
Giallongo A, Parisi GF, Papale M, Manti S, Mulé E, Aloisio D, Terlizzi V, Rotolo N, Leonardi S. Effects of Elexacaftor/Tezacaftor/Ivacaftor on Cardiorespiratory Polygraphy Parameters and Respiratory Muscle Strength in Cystic Fibrosis Patients with Severe Lung Disease. Genes. 2023; 14(2):449. https://doi.org/10.3390/genes14020449
Chicago/Turabian StyleGiallongo, Alessandro, Giuseppe Fabio Parisi, Maria Papale, Sara Manti, Enza Mulé, Donatella Aloisio, Vito Terlizzi, Novella Rotolo, and Salvatore Leonardi. 2023. "Effects of Elexacaftor/Tezacaftor/Ivacaftor on Cardiorespiratory Polygraphy Parameters and Respiratory Muscle Strength in Cystic Fibrosis Patients with Severe Lung Disease" Genes 14, no. 2: 449. https://doi.org/10.3390/genes14020449