
Candidate Genes and Gene Networks Change with Age in Japanese Black Cattle by Blood Transcriptome Analysis
 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Laboratory Animal and Sample Collection
2.2. RNA Extraction, Sequencing, and Data Analysis
2.3. Differentially Expressed Genes (DEGs) Analysis
2.4. Weighted Gene Co-Expression Network Analysis
2.5. Functional Enrichment and PPI Analysis
2.6. Exon-Wide Selection Signature
3. Results
3.1. Data Analysis of Transcriptome
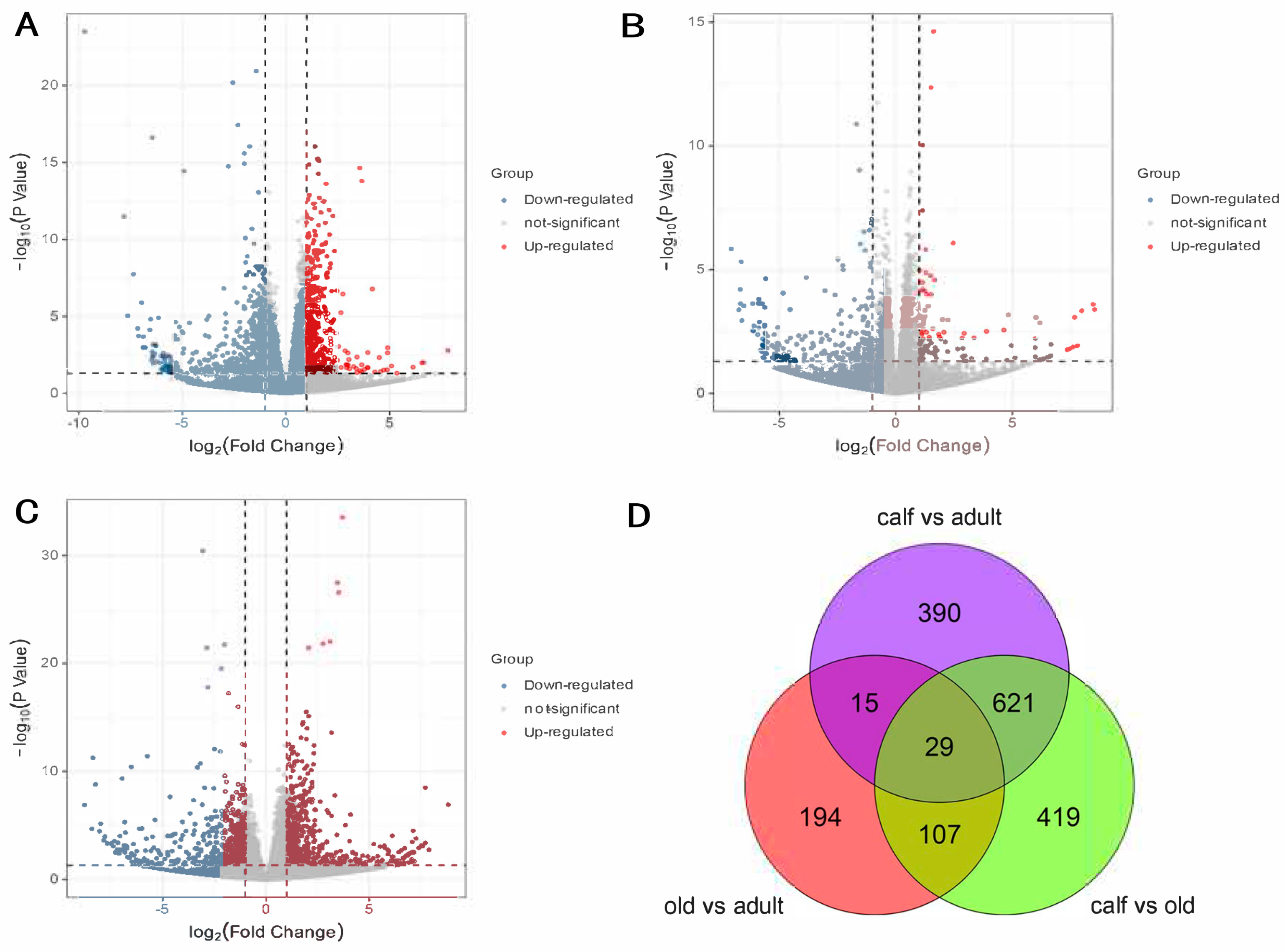
3.2. Differentially Expressed Genes across Three Age Stages
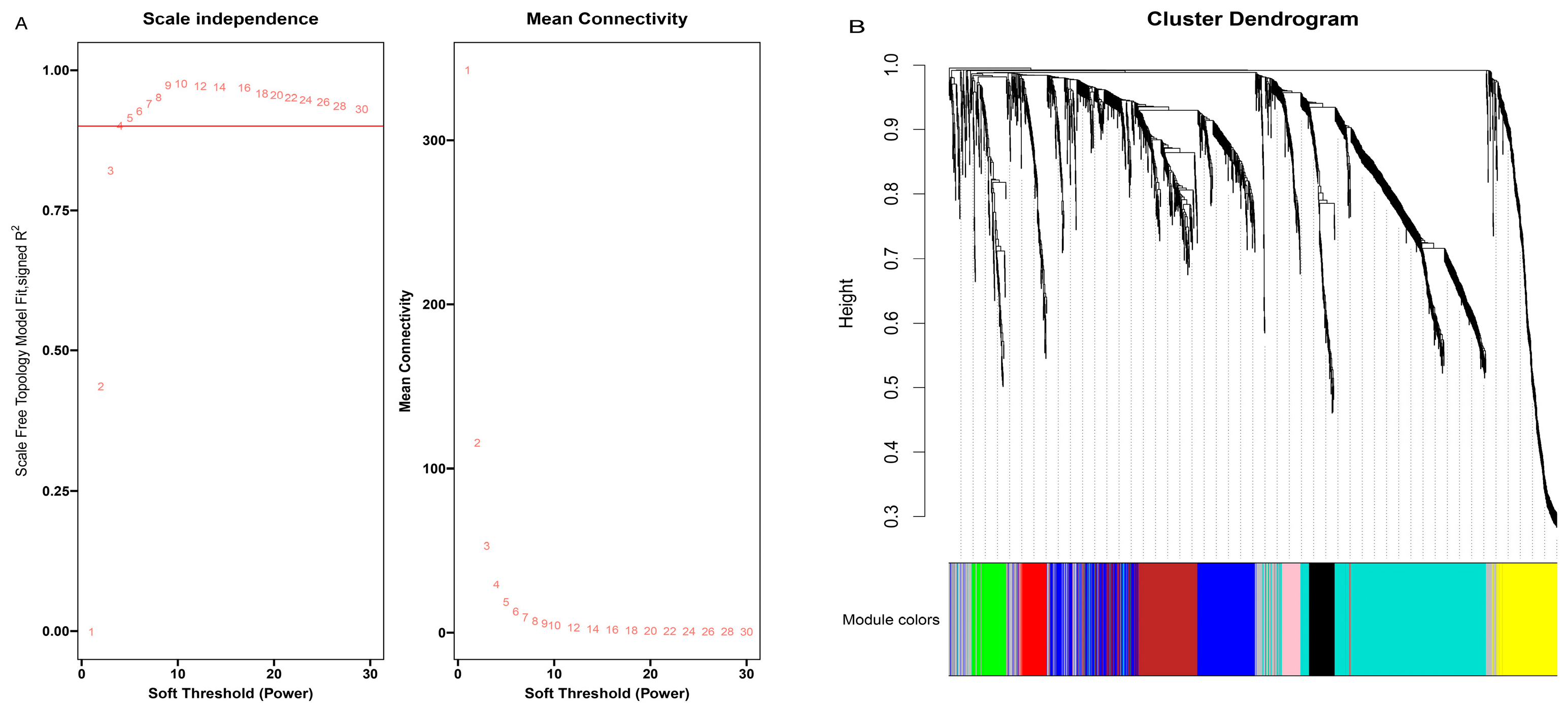
3.3. Construction of Weighted Gene Co-Expression Network and Module Detection
3.4. Identification of Specific Modules for Each Age Stage
3.5. Functional Enrichment of Stage-Specific Modules
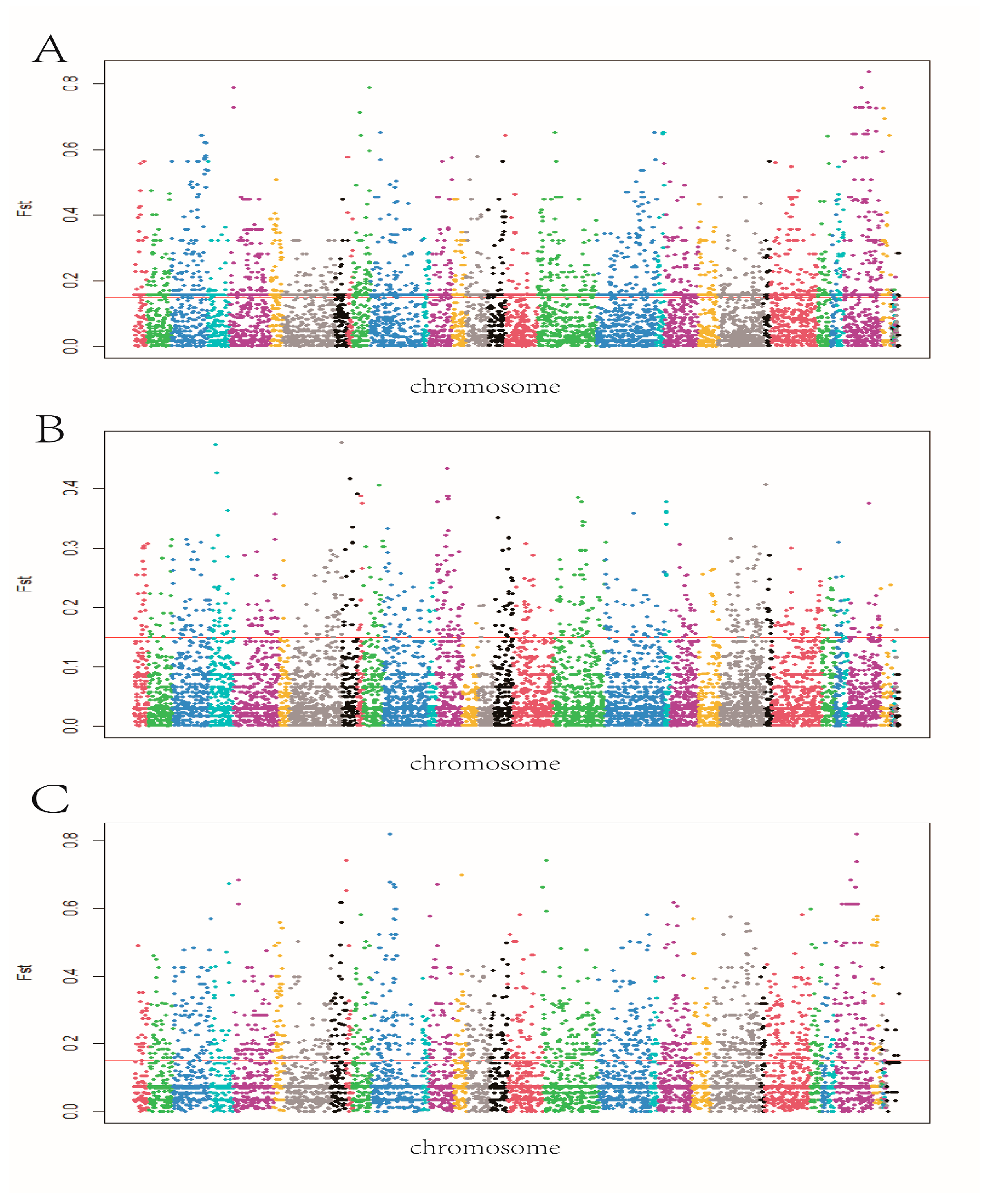
3.6. Exon-Wide Selection Signature
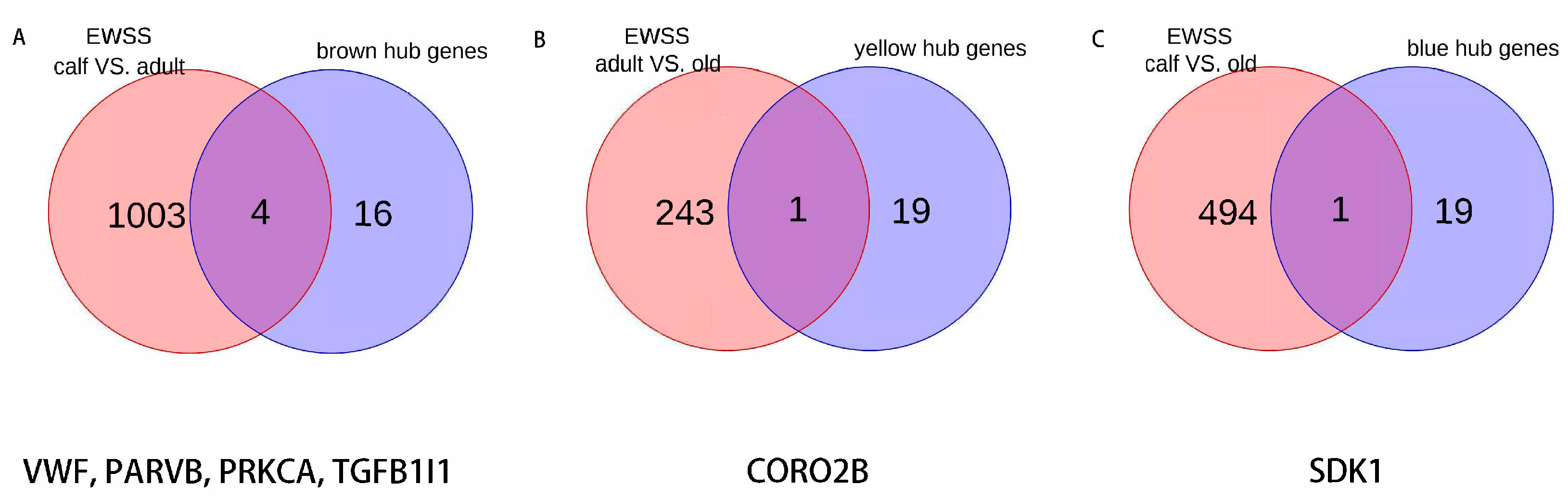
3.7. Candidate Genes for Each Age Stage
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chakraborty, D.; Sharma, N.; Kour, S.; Sodhi, S.S.; Gupta, M.K.; Lee, S.J.; Son, Y.O. Applications of Omics Technology for Livestock Selection and Improveme Nt. Front. Genet. 2022, 13, 774113. [Google Scholar] [CrossRef]
- Bickhart, D.M.; McClure, J.C.; Schnabel, R.D.; Rosen, B.D.; Medrano, J.F.; Smith, T.P.L. Symposium Review: Advances in Sequencing Technology Herald a New Front Ier in Cattle Genomics and Genome-Enabled Selection. J. Dairy Sci. 2020, 103, 5278–5290. [Google Scholar] [CrossRef]
- Inoue, K.; Inoue, Y.; Oe, T.; Nishimura, M. Genomic Imprinting Variances of Beef Carcass Traits and Physiochemical Characteristics in Japanese Black Cattle. Anim. Sci. J. 2021, 92, e13504. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, J.; Raza, S.H.A.; Deng, J.; Ma, J.; Qu, X.; Yu, S.; Zhang, D.; Alshammari, A.M.; Almohaimeed, H.M.; et al. Identification of the Hub Genes Related to Adipose Tissue Metabolism of Bovine. Front. Vet. Sci. 2022, 9, 1014286. [Google Scholar] [CrossRef]
- Gotoh, T.; Nishimura, T.; Kuchida, K.; Mannen, H. The Japanese Wagyu Beef Industry: Current Situation and Future Prospects–A Review. Asian-Australas. J. Anim. Sci. 2018, 31, 933–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, S.; Iwamoto, E.; Kato, Y.; Shinohara, M.; Shirai, Y.; Yamanoue, M. Comparative Metabolomics of Japanese Black Cattle Beef and Other Meats Using Gas Chromatography-Mass Spectrometry. Biosci. Biotechnol. Biochem. 2019, 83, 137–147. [Google Scholar] [CrossRef]
- Ueda, S.; Hosoda, M.; Yoshino, K.-I.; Yamanoue, M.; Shirai, Y. Gene Expression Analysis Provides New Insights into the Mechanism of Intramuscular Fat Formation in Japanese Black Cattle. Genes 2021, 12, 1107. [Google Scholar] [CrossRef] [PubMed]
- An, B.; Xia, J.; Chang, T.; Wang, X.; Xu, L.; Zhang, L.; Gao, X.; Chen, Y.; Li, J.; Gao, H. Genome-Wide Association Study Reveals Candidate Genes Associated with Body Measurement Traits in Chinese Wagyu Beef Cattle. Anim. Genet. 2019, 50, 386–390. [Google Scholar] [CrossRef]
- Sithyphone, K.; Yabe, M.; Horita, H.; Hayashi, K.; Fumita, T.; Shiotsuka, Y.; Etoh, T.; Ebara, F.; Samadmanivong, O.; Wegner, J.; et al. Comparison of Feeding Systems: Feed Cost, Palatability and Environmental Impact among Hay-Fattened Beef, Consistent Grass-Only-Fed Beef and Conventional Marbled Beef in Wagyu (Japanese Black Cattle). Anim. Sci. J. 2011, 82, 352–359. [Google Scholar] [CrossRef]
- Arishima, T.; Wakaguri, H.; Nakashima, R.; Sakakihara, S.; Kawashima, K.; Sugimoto, Y.; Suzuki, Y.; Sasaki, S. Comprehensive Analysis of 124 Transcriptomes from 31 Tissues in Developing, Juvenile, and Adult Japanese Black Cattle. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2022, 29, dsac032. [Google Scholar] [CrossRef]
- Chaussabel, D. Assessment of Immune Status Using Blood Transcriptomics and Potential Implications for Global Health. Semin. Immunol. 2015, 27, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamatha, P.; Ershler, W.; Longo, D. Bone Marrow, Thymus and Blood: Changes Across the Lifespan. Aging Health 2009, 5, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Liew, C.-C.; Ma, J.; Tang, H.-C.; Zheng, R.; Dempsey, A.A. The Peripheral Blood Transcriptome Dynamically Reflects System Wide Biology: A Potential Diagnostic Tool. J. Lab. Clin. Med. 2006, 147, 126–132. [Google Scholar] [CrossRef]
- Herder, C.; Karakas, M.; Koenig, W. Biomarkers for the Prediction of Type 2 Diabetes and Cardiovascular Disease. Clin. Pharmacol. Ther. 2011, 90, 52–66. [Google Scholar] [CrossRef]
- Bai, X.; Zheng, Z.; Liu, B.; Ji, X.; Bai, Y.; Zhang, W. Whole Blood Transcriptional Profiling Comparison between Different Mil k Yield of Chinese Holstein Cows Using RNA-Seq Data. BMC Genom. 2016, 17 (Suppl. 7), 512. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Tang, X.; Wu, M.; Li, Q.; Yi, X.; Liu, S.; Jiang, J.; Wang, S.; Sun, X. Transcriptome Characterization of Short Distance Transport Stress in Beef Cattle Blood. Front. Genet. 2021, 12, 616388. [Google Scholar] [CrossRef]
- Yang, Y.; Chang, C.; Baiyin, B.; Liu, Z.; Guo, L.; Zhou, L.; Liu, B.; Shi, C.; Zhang, W. Blood Transcriptome Analysis of Beef Cow with Different Parity Revealed Candidate Genes and Gene Networks Regulating the Postpartum Diseases. Genes 2022, 13, 1671. [Google Scholar] [CrossRef]
- Bongen, E.; Lucian, H.; Khatri, A.; Fragiadakis, G.K.; Bjornson, Z.B.; Nolan, G.P.; Utz, P.J.; Khatri, P. Sex Differences in the Blood Transcriptome Identify Robust Changes in Immune Cell Proportions with Aging and Influenza Infection. Cell Rep. 2019, 29, 1961–1973.e4. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Liu, Q.; Shen, F.; Fan, Z.; Hou, R.; Yue, B.; Zhang, X. Transcriptome Analysis Reveals Immune-Related Gene Expression Changes with Age in Giant Panda (Ailuropoda Melanoleuca) Blood. Aging 2019, 11, 249–262. [Google Scholar] [CrossRef]
- Lee, J.-R.; Choe, S.-H.; Kim, Y.-H.; Cho, H.-M.; Park, H.-R.; Lee, H.-E.; Jin, Y.B.; Kim, J.-S.; Jeong, K.J.; Park, S.-J.; et al. Longitudinal Profiling of the Blood Transcriptome in an African Green Monkey Aging Model. Aging 2020, 13, 846–864. [Google Scholar] [CrossRef]
- Nikolich-Žugich, J. The Twilight of Immunity: Emerging Concepts in Aging of the Immune System. Nat. Immunol. 2017, 19, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A Fast Spliced Aligner with Low Memory Requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Hall, S.J.G. Genetic Differentiation among Livestock Breeds—Values for Fst. Animals 2022, 12, 1115. [Google Scholar] [CrossRef]
- Vorobyeva, N.; Medvedev, I. Platelet Function Activity in Black-Motley Calves during the Dairy Phase. BIO Web Conf. 2020, 17, 00167. [Google Scholar] [CrossRef] [Green Version]
- Glagoleva, T.I.; Medvedev, I.N. Physiological Features of Aggregation of the Main Formed Elements of blood in Calves at the Beginning of Early Ontogenesis. BIO Web Conf. 2020, 17, 00161. [Google Scholar] [CrossRef] [Green Version]
- Oshurkova, J.L.; Medvedev, I.N. Functional Features of Platelets in Newborn Calves Ayrshire Breed. Dusunen Adam 2018, 9, 313–318. [Google Scholar]
- Vorobyeva, N.V.; Medvedev, I.N. Functional Platelet Activity in Dutch Newborn Calves. IOP Conf. Ser. Earth Environ. Sci. 2020, 421, 022042. [Google Scholar] [CrossRef]
- Balliu, B.; Durrant, M.; Goede, O.d.; Abell, N.; Li, X.; Liu, B.; Gloudemans, M.J.; Cook, N.L.; Smith, K.S.; Knowles, D.A.; et al. Genetic Regulation of Gene Expression and Splicing during a 10-Year Period of Human Aging. Genome Biol. 2019, 20, 230. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.J.; Joehanes, R.; Pilling, L.C.; Schurmann, C.; Conneely, K.N.; Powell, J.; Reinmaa, E.; Sutphin, G.L.; Zhernakova, A.; Schramm, K.; et al. The Transcriptional Landscape of Age in Human Peripheral Blood. Nat. Commun. 2015, 6, 8570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luykx, J.J.; Olde Loohuis, L.M.; Neeleman, M.; Strengman, E.; Bakker, S.C.; Lentjes, E.; Borgdorff, P.; van Dongen, E.P.A.; Bruins, P.; Kahn, R.S.; et al. Peripheral Blood Gene Expression Profiles Linked to Monoamine Metabolite Levels in Cerebrospinal Fluid. Transl. Psychiatry 2016, 6, e983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viñuela, A.; Brown, A.A.; Buil, A.; Tsai, P.-C.; Davies, M.N.; Bell, J.T.; Dermitzakis, E.T.; Spector, T.D.; Small, K.S. Age-Dependent Changes in Mean and Variance of Gene Expression across tissues in a Twin Cohort. Hum. Mol. Genet. 2018, 27, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, F.; Hu, H.; Bakshi, A.; Robinson, M.R.; Powell, J.E.; Montgomery, G.W.; Goddard, M.E.; Wray, N.R.; Visscher, P.M.; et al. Integration of Summary Data from GWAS and EQTL Studies Predicts Complex Trait Gene Targets. Nat. Genet. 2016, 48, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Gilad, Y.; Rifkin, S.A.; Pritchard, J.K. Revealing the Architecture of Gene Regulation: The Promise of EQTL Studies. Trends Genet. 2008, 24, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, P.A. Cell Signaling: An Overview; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Li, S.; Cui, Y.; Li, M.; Zhang, W.; Sun, X.; Xin, Z.; Li, J. Acteoside Derived from Cistanche Improves Glucocorticoid-Induced Osteoporosis by Activating PI3K/AKT/MTOR Pathway. J. Investig. Surg. Off. J. Acad. Surg. Res. 2023, 36, 2154578. [Google Scholar] [CrossRef]
- Diniz, W.J.S.; Crouse, M.S.; Cushman, R.A.; McLean, K.J.; Caton, J.S.; Dahlen, C.R.; Reynolds, L.P.; Ward, A.K. Cerebrum, Liver, and Muscle Regulatory Networks Uncover Maternal Nutrition Effects in Developmental Programming of Beef Cattle during Early Pregnancy. Sci. Rep. 2021, 11, 2771. [Google Scholar] [CrossRef]
- Song, C.; Huang, Y.; Yang, Z.; Ma, Y.; Chaogetu, B.; Zhuoma, Z.; Chen, H. RNA-Seq Analysis Identifies Differentially Expressed Genes in Subcutaneous Adipose Tissue in Qaidaford Cattle, Cattle-Yak, and Angus Cattle. Animals 2019, 9, 1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Pan, Y.; Yang, L.; Xie, Y.; Chen, X.; Chang, J.; Hao, W.; Zhu, L.; Wan, B. Developmental Toxicity of TCBPA on the Nervous and Cardiovascular Syst Ems of Zebrafish (Danio Rerio): A Combination of Transcriptomic and Metabolomics. J. Environ. Sci. 2023, 127, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Taye, M.; Yoon, J.; Dessie, T.; Cho, S.; Oh, S.J.; Lee, H.-K.; Kim, H. Deciphering Signature of Selection Affecting Beef Quality Traits in Angus Cattle. Genes Genom. 2018, 40, 63–75. [Google Scholar] [CrossRef]
- Wu, P.; Zhang, X.; Zhang, G.; Chen, F.; He, M.; Zhang, T.; Wang, J.; Xie, K.; Dai, G. Transcriptome for the Breast Muscle of Jinghai Yellow Chicken at Early Growth Stages. PeerJ 2020, 8, e8950. [Google Scholar] [CrossRef] [Green Version]
- Díaz Galván, C.; Méndez Olvera, E.T.; Martínez Gómez, D.; Gloria Trujillo, A.; Hernández García, P.A.; Espinosa Ayala, E.; Palacios Martínez, M.; Lara Bueno, A.; Mendoza Martínez, G.D.; Velázquez Cruz, L.A. Influence of a Polyherbal Mixture in Dairy Calves: Growth Performance and Gene Expression. Front. Vet. Sci. 2021, 7, 623710. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared Principles in NF-KappaB Signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Chen, Z.J. Regulation of NF-ΚB by Ubiquitination. Curr. Opin. Immunol. 2013, 25, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Perkins, N.D. Post-Translational Modifications Regulating the Activity and Function of the Nuclear Factor Kappa B Pathway. Oncogene 2006, 25, 6717–6730. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The Master Regulator of Fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Periyasamy, S.; Sánchez, E.R. Antagonism of Glucocorticoid Receptor Transactivity and Cell Growth inhibition by Transforming Growth Factor-Beta through AP-1-Mediated Transcriptional Repression. Int. J. Biochem. Cell Biol. 2002, 34, 1571–1585. [Google Scholar] [CrossRef]
- Hatzirodos, N.; Hummitzsch, K.; Irving-Rodgers, H.F.; Breen, J.; Perry, V.E.A.; Anderson, R.A.; Rodgers, R.J. Transcript Abundance of Stromal and Thecal Cell Related Genes during bovine Ovarian Development. PLoS ONE 2019, 14, e0213575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandre, P.A.; Naval-Sanchez, M.; Porto-Neto, L.R.; Ferraz, J.B.S.; Reverter, A.; Fukumasu, H. Systems Biology Reveals NR2F6 and TGFB1 as Key Regulator s of Feed Efficiency in Beef Cattle. Front. Genet. 2019, 10, 230. [Google Scholar] [CrossRef] [PubMed]
- de Araujo Neto, F.R.; Santos, D.J.d.A.; Fernandes Júnior, G.A.; Aspilcueta-Borquis, R.R.; Nascimento, A.V.d.; de Oliveira Seno, L.; Tonhati, H.; de Oliveira, H.N. Genome-Wide Association Studies for Growth Traits in Buffaloes Using the Single Step Genomic BLUP. J. Appl. Genet. 2020, 61, 113–115. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, C.A.; Donaldson, D.I. PRKCA Polymorphism Changes the Neural Basis of Episodic Remembering in Healthy Individuals. PLoS ONE 2014, 9, e98018. [Google Scholar] [CrossRef]
- Bogard, A.S.; Tavalin, S.J. Protein Kinase C (PKC)ζPseudosubstrate Inhibitor Peptide Promiscuously Binds PKC Family Isoforms and Disrupts Conventional PKC Targeting and Translocation. Mol. Pharmacol. 2015, 88, 728–735. [Google Scholar] [CrossRef] [Green Version]
- Suzuma, K. AGE and PKC. Nihon Rinsho Jpn. J. Clin. Med. 2010, 68 (Suppl. 9), 246–249. [Google Scholar]
- Wang, H.; Zhong, J.; Zhang, C.; Chai, Z.; Cao, H.; Wang, J.; Zhu, J.; Wang, J.; Ji, Q. The Whole-Transcriptome Landscape of Muscle and Adipose Tissues Reveals the CeRNA Regulation Network Related to Intramuscular Fat Deposition in Yak. BMC Genom. 2020, 21, 347. [Google Scholar] [CrossRef]
- Dou, D.; Shen, L.; Zhou, J.; Cao, Z.; Luan, P.; Li, Y.; Xiao, F.; Guo, H.; Li, H.; Zhang, H. Genome-Wide Association Studies for Growth Traits in Broilers. BMC Genom. Data 2022, 23, 1. [Google Scholar] [CrossRef]
- Wu, C.-F.; Ng, K.-F.; Chen, C.-S.; Chang, P.-L.; Chuang, C.-K.; Weng, W.-H.; Liao, S.-K.; Pang, S.-T. Expression of Parvin-Beta Is a Prognostic Factor for Patients with Urothelial Cell Carcinoma of the Upper Urinary Tract. Br. J. Cancer 2010, 103, 852–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eslami, A.; Miyaguchi, K.; Mogushi, K.; Watanabe, H.; Okada, N.; Shibuya, H.; Mizushima, H.; Miura, M.; Tanaka, H. PARVB Overexpression Increases Cell Migration Capability and Defines high Risk for Endophytic Growth and Metastasis in Tongue Squamous Cell Carcinoma. Br. J. Cancer 2015, 112, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Fedarko, N.S. The Biology of Aging and Frailty. Clin. Geriatr. Med. 2011, 27, 27–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niccoli, T.; Partridge, L. Ageing as a Risk Factor for Disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamczyk, M.R.; Nevado, R.M.; Barettino, A.; Fuster, V.; Andrés, V. Biological Versus Chronological Aging. J. Am. Coll. Cardiol. 2020, 75, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic Regulation of Ageing: Linking Environmental Inputs to Genomic Stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, A.; Möller-Hackbarth, K.; Ebarasi, L.; Unnersjö Jess, D.; Zambrano, S.; Blom, H.; Wernerson, A.; Lal, M.; Patrakka, J. Coro2b, a Podocyte Protein Downregulated in Human Diabetic Nephropathy, Is Involved in the Development of Protamine Sulphate-Induced Foot Process Effacement. Sci. Rep. 2019, 9, 8888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, P.; Yang, X.; Li, Y.; Huang, B.; Cai, C. The Distribution of a Missense Mutation in SDK1 Gene across Native Chinese Breeds. Anim. Biotechnol. 2021, 3, 1–4. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, C.; Yang, Y.; Zhou, L.; Baiyin, B.; Liu, Z.; Guo, L.; Ma, F.; Wang, J.; Chai, Y.; Shi, C.; et al. Candidate Genes and Gene Networks Change with Age in Japanese Black Cattle by Blood Transcriptome Analysis. Genes 2023, 14, 504. https://doi.org/10.3390/genes14020504
Chang C, Yang Y, Zhou L, Baiyin B, Liu Z, Guo L, Ma F, Wang J, Chai Y, Shi C, et al. Candidate Genes and Gene Networks Change with Age in Japanese Black Cattle by Blood Transcriptome Analysis. Genes. 2023; 14(2):504. https://doi.org/10.3390/genes14020504
Chicago/Turabian StyleChang, Chencheng, Yanda Yang, Le Zhou, Batu Baiyin, Zaixia Liu, Lili Guo, Fengying Ma, Jie Wang, Yuan Chai, Caixia Shi, and et al. 2023. "Candidate Genes and Gene Networks Change with Age in Japanese Black Cattle by Blood Transcriptome Analysis" Genes 14, no. 2: 504. https://doi.org/10.3390/genes14020504