
Clinical and Genetic Features of Korean Patients with Achromatopsia

 , and
, and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patient Selection
2.2. Ophalmic Examinations
2.3. Genotyping
2.4. Statistical Analysis
3. Results
3.1. Demographics and Clinical Presentation
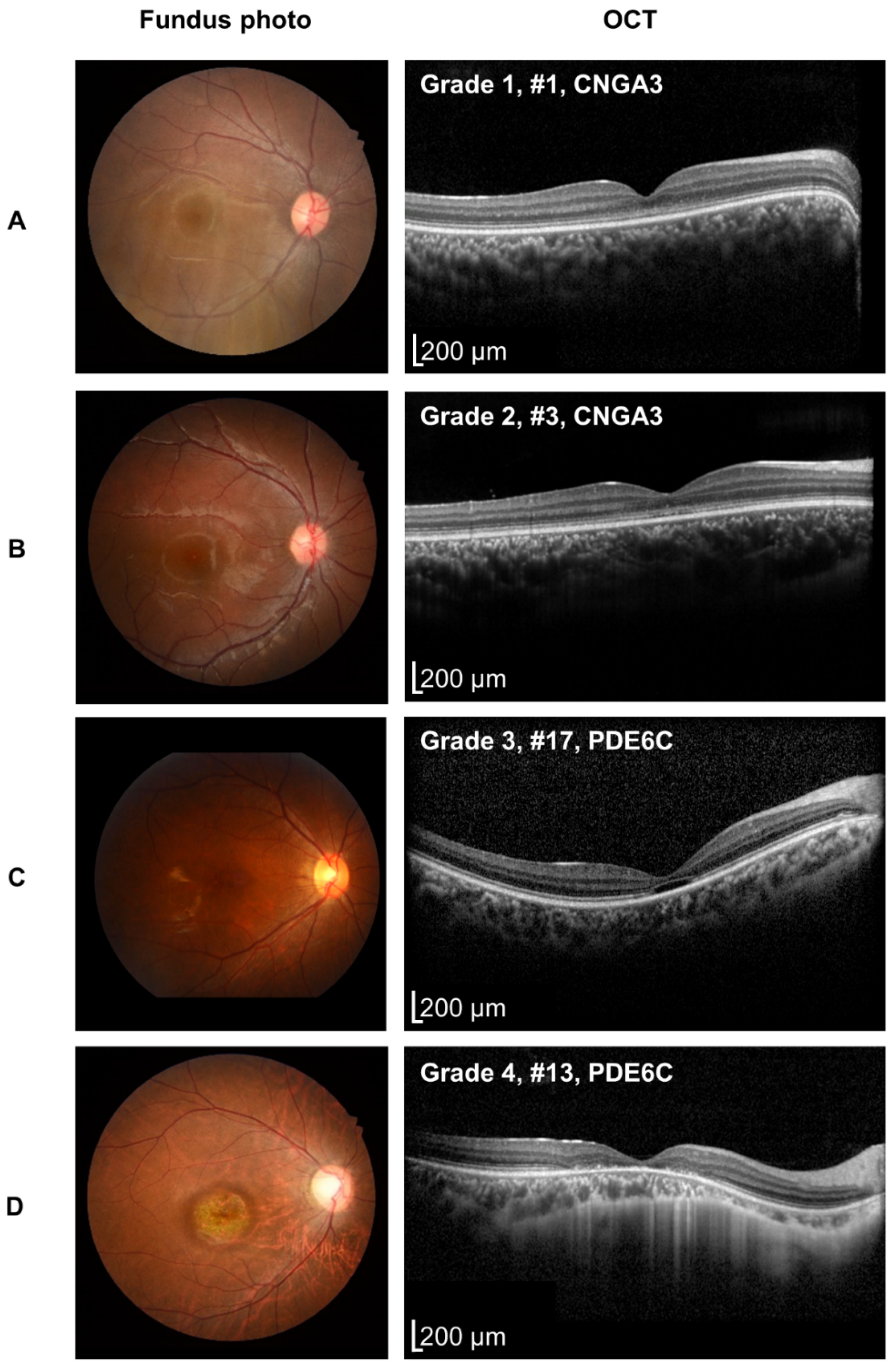
3.2. Retinal Phenotypes
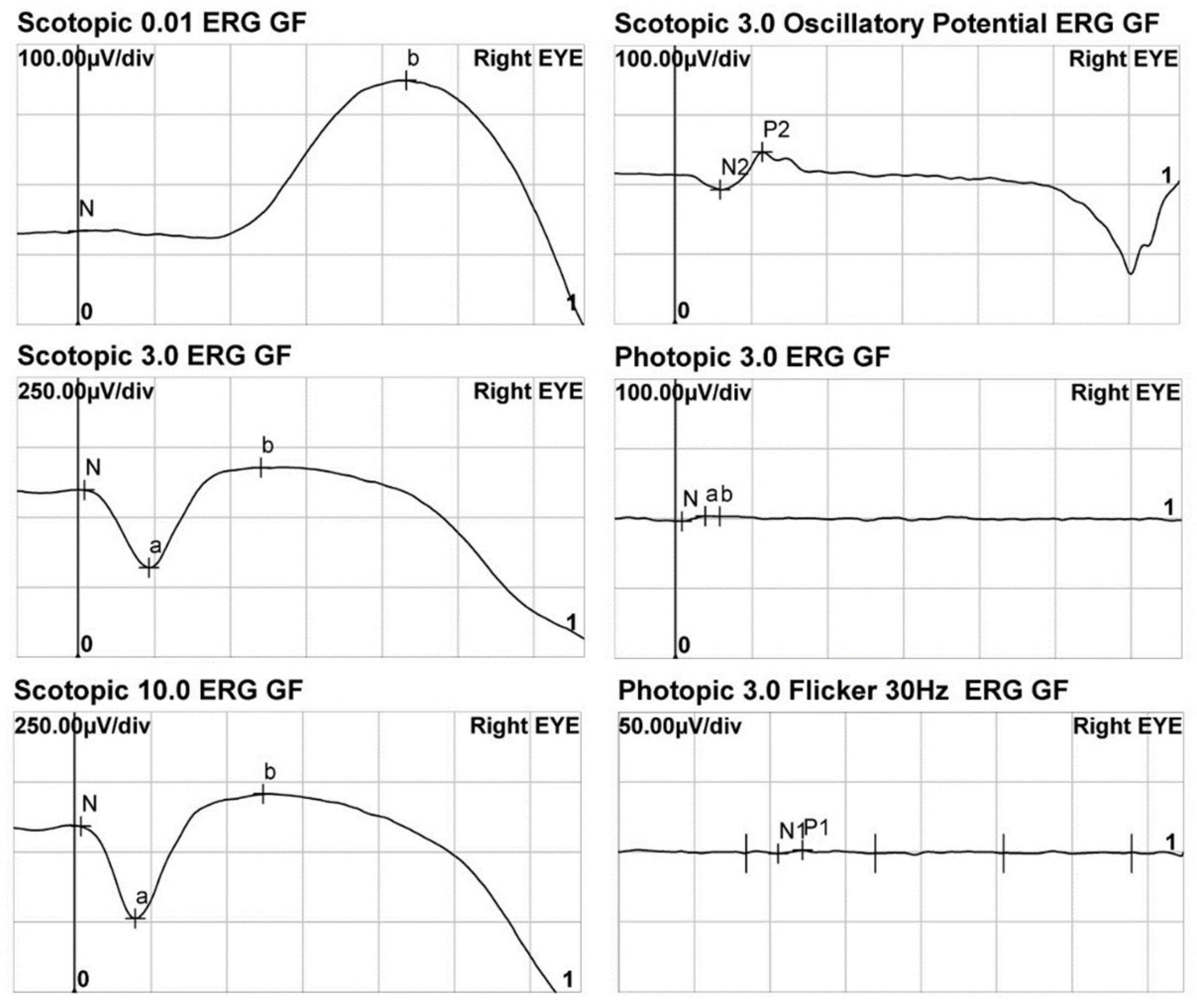
3.3. Electroretinogram
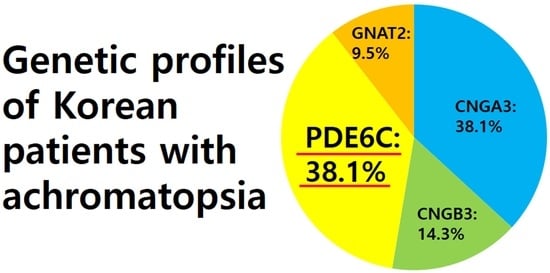
3.4. Genotypes
3.5. Correlations of Genotypes and Phenotypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sundaram, V.; Wilde, C.; Aboshiha, J.; Cowing, J.; Han, C.; Langlo, C.S.; Chana, R.; Davidson, A.E.; Sergouniotis, P.I.; Bainbridge, J.W.; et al. Retinal structure and function in achromatopsia: Implications for gene therapy. Ophthalmology 2014, 121, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalakis, S.; Gerhardt, M.; Rudolph, G.; Priglinger, S.; Priglinger, C. Achromatopsia: Genetics and Gene Therapy. Mol. Diagn. Ther. 2022, 26, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Cehajic-Kapetanovic, J.; Singh, M.S.; Zrenner, E.; MacLaren, R.E. Bioengineering strategies for restoring vision. Nat. Biomed. Eng. 2022. [Google Scholar] [CrossRef] [PubMed]
- Brunetti-Pierri, R.; Karali, M.; Melillo, P.; Di Iorio, V.; De Benedictis, A.; Iaccarino, G.; Testa, F.; Banfi, S.; Simonelli, F. Clinical and Molecular Characterization of Achromatopsia Patients: A Longitudinal Study. Int. J. Mol. Sci. 2021, 22, 1681. [Google Scholar] [CrossRef] [PubMed]
- Ouechtati, F.; Merdassi, A.; Bouyacoub, Y.; Largueche, L.; Derouiche, K.; Ouragini, H.; Nouira, S.; Tiab, L.; Baklouti, K.; Rebai, A.; et al. Clinical and genetic investigation of a large Tunisian family with complete achromatopsia: Identification of a new nonsense mutation in GNAT2 gene. J. Hum. Genet. 2011, 56, 22–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saqib, M.A.; Awan, B.M.; Sarfraz, M.; Khan, M.N.; Rashid, S.; Ansar, M. Genetic analysis of four Pakistani families with achromatopsia and a novel S4 motif mutation of CNGA3. Jpn. J. Ophthalmol. 2011, 55, 676–680. [Google Scholar] [CrossRef]
- Sun, W.; Li, S.; Xiao, X.; Wang, P.; Zhang, Q. Genotypes and phenotypes of genes associated with achromatopsia: A reference for clinical genetic testing. Mol. Vis. 2020, 26, 588–602. [Google Scholar]
- Andreasson, S.; Tornqvist, K. Electroretinograms in patients with achromatopsia. Acta Ophthalmol. 1991, 69, 711–716. [Google Scholar] [CrossRef]
- Georgiou, M.; Robson, A.G.; Singh, N.; Pontikos, N.; Kane, T.; Hirji, N.; Ripamonti, C.; Rotsos, T.; Dubra, A.; Kalitzeos, A.; et al. Deep Phenotyping of PDE6C-Associated Achromatopsia. Investig. Ophthalmol. Vis. Sci 2019, 60, 5112–5123. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, J.P.; Sherman, J.; Zweifel, S.A.; Chen, R.W.; Duncker, T.; Kohl, S.; Baumann, B.; Wissinger, B.; Yannuzzi, L.A.; Tsang, S.H. Spectral-domain optical coherence tomography staging and autofluorescence imaging in achromatopsia. JAMA Ophthalmol. 2014, 132, 437–445. [Google Scholar] [CrossRef]
- Scoles, D.; Flatter, J.A.; Cooper, R.F.; Langlo, C.S.; Robison, S.; Neitz, M.; Weinberg, D.V.; Pennesi, M.E.; Han, D.P.; Dubra, A.; et al. Assessing Photoreceptor Structure Associated with Ellipsoid Zone Disruptions Visualized with Optical Coherence Tomography. Retina 2016, 36, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohl, S.; Jägle, H.; Wissinger, B.; Zobor, D. Achromatopsia. In GeneReviews(®); Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Marmor, M.F.; Fulton, A.B.; Holder, G.E.; Miyake, Y.; Brigell, M.; Bach, M.; International Society for Clinical Electrophysiology of Vision. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc. Ophthalmol. 2009, 118, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.W.; Joo, K.; Woo, S.J. Clinical Characterization of Korean Patients with Pseudoxanthoma Elasticum and Angioid Streaks. Genes 2021, 12, 1207. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Plagnol, V.; Curtis, J.; Epstein, M.; Mok, K.Y.; Stebbings, E.; Grigoriadou, S.; Wood, N.W.; Hambleton, S.; Burns, S.O.; Thrasher, A.J.; et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012, 28, 2747–2754. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.T.; Han, J. Missed Heterozygous Deletion in Study of Next-Generation Sequencing for Molecular Diagnosis in Patients With Infantile Nystagmus Syndrome. JAMA Ophthalmol. 2019, 137, 1465–1466. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Huang, L.; Xiao, X.; Jia, X.; Guo, X.; Zhang, Q. Identification of CNGA3 mutations in 46 families: Common cause of achromatopsia and cone-rod dystrophies in Chinese patients. JAMA Ophthalmol. 2014, 132, 1076–1083. [Google Scholar] [CrossRef] [Green Version]
- Wissinger, B.; Gamer, D.; Jägle, H.; Giorda, R.; Marx, T.; Mayer, S.; Tippmann, S.; Broghammer, M.; Jurklies, B.; Rosenberg, T.; et al. CNGA3 Mutations in Hereditary Cone Photoreceptor Disorders. Am. J. Hum. Genet. 2001, 69, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Rim, J.H.; Lee, S.T.; Gee, H.Y.; Lee, B.J.; Choi, J.R.; Park, H.W.; Han, S.H.; Han, J. Accuracy of Next-Generation Sequencing for Molecular Diagnosis in Patients With Infantile Nystagmus Syndrome. JAMA Ophthalmol. 2017, 135, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Michaelides, M.; Aligianis, I.A.; Ainsworth, J.R.; Mollon, J.D.; Maher, E.R.; Moore, A.T.; Hunt, D.M. Achromatopsia caused by novel mutations in both CNGA3 and CNGB3. J. Med. Genet. 2004, 41, e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Joo, K.; Seong, M.W.; Kim, M.J.; Park, K.H.; Park, S.S.; Woo, S.J. Genetic Mutation Profiles in Korean Patients with Inherited Retinal Diseases. J. Korean Med. Sci. 2019, 34, e161. [Google Scholar] [CrossRef]
- Kohl, S.; Marx, T.; Giddings, I.; Jägle, H.; Jacobson, S.G.; Apfelstedt-Sylla, E.; Zrenner, E.; Sharpe, L.T.; Wissinger, B. Total colourblindness is caused by mutations in the gene encoding the α-subunit of the cone photoreceptor cGMP-gated cation channel. Nat. Genet. 1998, 19, 257–259. [Google Scholar] [CrossRef]
- Ahuja, Y.; Kohl, S.; Traboulsi, E. CNGA3 mutations in two United Arab Emirates families with chromatopsia. Mol. Vis. 2008, 10, 1293–1297. [Google Scholar]
- Mayer, A.K.; Van Cauwenbergh, C.; Rother, C.; Baumann, B.; Reuter, P.; De Baere, E.; Wissinger, B.; Kohl, S.; Group, A.S. CNGB3 mutation spectrum including copy number variations in 552 achromatopsia patients. Hum. Mutat. 2017, 38, 1579–1591. [Google Scholar] [CrossRef]
- Huang, L.; Xiao, X.; Li, S.; Jia, X.; Wang, P.; Sun, W.; Xu, Y.; Xin, W.; Guo, X.; Zhang, Q. Molecular genetics of cone-rod dystrophy in Chinese patients: New data from 61 probands and mutation overview of 163 probands. Exp. Eye Res. 2016, 146, 252–258. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. ClinVar; [VCV000970700.5]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000970700.5 (accessed on 27 January 2023).
- Thiadens, A.A.; den Hollander, A.I.; Roosing, S.; Nabuurs, S.B.; Zekveld-Vroon, R.C.; Collin, R.W.; De Baere, E.; Koenekoop, R.K.; van Schooneveld, M.J.; Strom, T.M.; et al. Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am. J. Hum. Genet. 2009, 85, 240–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abouelhoda, M.; Sobahy, T.; El-Kalioby, M.; Patel, N.; Shamseldin, H.; Monies, D.; Al-Tassan, N.; Ramzan, K.; Imtiaz, F.; Shaheen, R.; et al. Clinical genomics can facilitate countrywide estimation of autosomal recessive disease burden. Genet. Med. 2016, 18, 1244–1249. [Google Scholar] [CrossRef] [Green Version]
- National Center for Biotechnology Information. ClinVar. [VCV001213877.1]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV001213877.1 (accessed on 27 January 2023).
- National Center for Biotechnology Information. ClinVar. [VCV001017583.2]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV001017583.2 (accessed on 27 January 2023).
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef]
- Thiadens, A.A.; Slingerland, N.W.; Roosing, S.; van Schooneveld, M.J.; van Lith-Verhoeven, J.J.; van Moll-Ramirez, N.; van den Born, L.I.; Hoyng, C.B.; Cremers, F.P.; Klaver, C.C. Genetic etiology and clinical consequences of complete and incomplete achromatopsia. Ophthalmology 2009, 116, 1984–1989 e1981. [Google Scholar] [CrossRef]
- Hirji, N.; Aboshiha, J.; Georgiou, M.; Bainbridge, J.; Michaelides, M. Achromatopsia: Clinical features, molecular genetics, animal models and therapeutic options. Ophthalmic Genet. 2018, 39, 149–157. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Sex | Age (Years) | Follow-Up Duration (Years) | Visual Acuity at Baseline (LogMAR) | Visual Acuity at Last Visit (LogMAR) | Genotype | OCT Grade at Last Examinations | Foveal Hypoplasia on OCT | ERG Rod Responses | ERG Cone Responses | Color Vision Test |
|---|---|---|---|---|---|---|---|---|---|---|---|
| #1 | M | 0.6 | 14.4 | 1.3/1.0 | 1.0/1.0 | CNGA3 | 1 | No | Normal | NM | Color blindness |
| #2 | F | 1.2 | 6.2 | 1.4/1.4 | 0.82/1.0 | CNGA3 | 1 | Yes | Not conducted | Not conducted | Not conducted |
| #3 | M | 1.5 | 10.6 | 0.7/0.82 | 0.7/0.7 | CNGA3 | 2 | No | Present | NM | Color blindness |
| #4 | M | 3.4 | 13.8 | 0.52/0.52 | 0.82/1.0 | CNGA3 | 2 | Yes | Decreased | NM | Not conducted |
| #5 | M | 2.5 | 6.2 | 0.7/1.0 | 1.0/1.0 | CNGA3 | 1 | No | Present | NM | Color blindness |
| #6 | M | 8 | 4.4 | 1.3/1.3 | 1.1/1.3 | CNGA3 | 1 | Yes | Present | NM | Not conducted |
| #7 | M | 12.4 | 0 | 0.7/0.7 | 0.7/0.7 | CNGA3 | 1 | No | Not conducted | Not conducted | Not conducted |
| #8 | F | 30.7 | 1 | 0.82/0.82 | 0.82/0.82 | CNGA3 | 2 | No | Not conducted | Not conducted | Color blindness |
| #9 | F | 0.5 | 14.9 | 1.3/1.3 | 1.4/1.4 | CNGB3 | 3 | Yes | Present | NM | Not conducted |
| #10 | F | 2.6 | 0.5 | 1.22/1.0 | 1.22/1.0 | CNGB3 | Not conducted | Not conducted | Present | NM | Not conducted |
| #11 | M | 38.2 | 1.8 | 0.82/1.0 | 0.82/1.0 | CNGB3 | 3 | Yes | Decreased | NM | Not conducted |
| #12 | M | 0.6 | 8.5 | 1.0/1.0 | 0.82/0.82 | PDE6C | 2 | Yes | Present | NM | Color blindness |
| #13 | M | 0.6 | 25 | 1.0/1.0 | 1.0/1.0 | PDE6C | 4 | No | Decreased | NM | Not conducted |
| #14 | M | 4.3 | 3.3 | 1.0/1.3 | 1.1/1.0 | PDE6C | 2 | No | Normal | NM | Color blindness |
| #15 | F | 12.3 | 0.4 | 0.82/0.82 | 0.82/0.82 | PDE6C | 2 | No | Present | NM | Not conducted |
| #16 | F | 13 | 14.2 | 1.3/1.3 | 1.0/1.0 | PDE6C | 4 | No | Decreased | NM | Color blindness |
| #17 | F | 11.3 | 14.7 | 1.0/0.82 | 1.0/1.0 | PDE6C | 3 | No | Present | NM | Not conducted |
| #18 | M | 16.2 | 2 | 0.82/0.82 | 0.82/0.82 | PDE6C | 3 | No | Present | NM | Color blindness |
| #19 | M | 47.2 | 0 | Not conducted | Not conducted | PDE6C | 4 | No | Not conducted | Not conducted | Not conducted |
| #20 | M | 9.7 | 2.4 | 1.52/1.52 | 1.4/1.4 | GNAT2 | 1 | No | Present | NM | Color blindness |
| #21 | F | 11.9 | 10 | 1.0/1.0 | 1.0/0.82 | GNAT2 | 1 | No | Present | NM | N/A |
| No. | DA 0.01 b-Wave Amplitude | LA 3.0 | LA 30 Hz | |||||
|---|---|---|---|---|---|---|---|---|
| a-Wave Amplitude | b-Wave Amplitude | N1-P1 Amplitude | ||||||
| RE | LE | RE | LE | RE | LE | RE | LE | |
| #1 | 215 | 196 | 7.54 | 7.93 | 0.485 | 3.24 | NM | NM |
| #3 | 38 | 36.5 | NM | NM | NM | NM | NM | NM |
| #4 | 40.8 | 69.4 | NM | NM | NM | NM | NM | NM |
| #5 | 46.6 | 64.5 | NM | NM | NM | NM | NM | NM |
| #6 | 37 | 40.6 | Not conducted | Not conducted | Not conducted | Not conducted | NM | NM |
| #9 | 33.3 | 28.8 | NM | NM | NM | NM | NM | NM |
| #10 | 41.3 | 8.3 | NM | NM | NM | NM | NM | NM |
| #11 | 44.9 | 62.7 | NM | NM | NM | NM | NM | NM |
| #12 | 44.0 | 31.1 | NM | NM | NM | NM | NM | NM |
| #13 | 67.1 | 93.5 | 7.0 | 3.0 | NM | NM | NM | NM |
| #14 | NM | NM | NM | NM | NM | NM | NM | NM |
| #15 | 19.4 | 19.5 | NM | NM | NM | NM | NM | NM |
| #16 | 85.1 | 77.9 | NM | NM | NM | NM | 3.64 | 1.62 |
| #17 | 183.9 | 181.7 | NM | NM | NM | NM | NM | NM |
| #18 | 34.8 | 35.2 | NM | NM | NM | NM | NM | NM |
| #20 | 7.9 | 31.1 | NM | NM | NM | NM | NM | NM |
| #21 | 176.3 | 134.8 | NM | NM | NM | NM | NM | NM |
| No. | Gene | Nucleotide Variation | Protein Variation | Zygosity | CADD PHRED Score (GRCh37-v1.6) | Allele Frequency (%) in gnomAD | Pathogenicity (ACMG Classification and Its Criteria) | Previously Reported |
|---|---|---|---|---|---|---|---|---|
| #1 | CNGA3 | c.2T>A c.1001C>T | p.(Met1?) p.(Ser334Phe) | heterozygous | 22.8 26 | P (PVS1, PM2, PP3) LP | Novel [20] | |
| #2 | CNGA3 | c.829C>T c.1001C>T | p.(Arg277Cys) p.(Ser334Phe) | Compound heterozygous | 31 26 | 25/251350 | P LP | [21] [20] |
| #3 | CNGA3 | c.553C>G c.1190G>T | p.(Leu185Val) p.(Gly397Val) | Compound heterozygous | 20.5 25.7 | LP LP | [22] [23] | |
| #4 | CNGA3 | c.553C>G c.848G>A | p.(Leu185Val) p.(Arg283Gln) | Heterozygous | 20.5 28.8 | 1/251394 16/251352 | LP P | [22] [21] |
| #5 | CNGA3 | c.1262del c.1642G>A | p.(Lys421Serfs*44) p.(Gly548Arg) | Compound heterozygous | 32 28.1 | 6/251292 | P P | [24] [25] |
| #6 | CNGA3 | c.553C>T c.847C>T | p.(Leu185Val) p.(Arg283Trp) | Heterozygous | 20.5 26.5 | 1/251394 25/251318 | LP LP | [22] [26] |
| #7 | CNGA3 | c.829C>T | p.(Arg277Cys) | Homozygous | 31 | 24/251350 | P | [21] |
| #8 | CNGA3 | c.158_161dup c.1190G>T | p.(Arg55Aspfs*6) p.(Gly397Val) | Heterozygous | 24 25.7 | LP (PVS1, PM2) LP | Novel [22,23] | |
| #9 | CNGB3 | c.1928+2T>C | Homozygous | 33 | P | [24] | ||
| #10 | CNGB3 | c.1258_1277del c.1579-2A>G | p.(Ile420Phefs*35) | Compound heterozygous | 33 33 | 1/249908 | P (PVS1, PP1) P | Novel [27] |
| #11 | CNGB3 | c.1579-2A>G Exon 16 deletion | Heterozygous | 33 | 1/249908 | P LP (PVS1, PM2) | [27] Novel | |
| #12 | PDE6C | c.1771G>A c.2269C>T | p.(Glu591Lys) p.(Gln757*) | Compound heterozygous | 28.1 50 | LP LP (PVS1, PM2) | [28] Novel | |
| #13 | PDE6C | c.1646T>C c.1766A>G | p.(Met549Thr) p.(Asp589Gly) | Compound heterozygous | 22.5 24.6 | 2/251386 | LP US | [8] [29] |
| #14 | PDE6C | c.85C>T c.712C>T | p.(Arg29Trp) p.(Arg238*) | Heterozygous | 24.1 36 | 6/277228 2/246146 | P P | [30] [31] |
| #15 | PDE6C | c.480G>T Exon 1 deletion | p.(Lys160Asn) | Compound heterozygous | 33 | 1/245444 | P P (PVS1, PM2, PP3) | [32] Novel |
| #16 | PDE6C | c.85C>T c.1771G>A | p.(Arg29Trp) p.(Glu591Lys) | Compound heterozygous | 24.1 28.1 | 6/282886 2/251120 | P LP | [30] [28] |
| #17 | PDE6C | c.1643G>T c.2507delG | p.(Trp548Leu) p.(Gly836Glufs*21) | Heterozygous | 31 26.1 | 2/251354 | LP (PVS1, PP1) P (PS3, PM1, PM2, PM3, PP3) | Novel Novel |
| #18 | PDE6C | c.85C>T c.827G>A | p.(Arg29Trp) p.(Arg276Gln) | Compound heterozygous | 24.1 26.7 | 6/282886 5/282768 | P LP | [30] [33] |
| #19 | PDE6C | c.1540T>A c.1785G>A | p.(Phe514Ile) p.(Met595Ile) | Heterozygous | 21.3 27.8 | US (PM1, PM2, PM3) US (PM2, PM3, PP3) | Novel Novel | |
| #20 | GNAT2 | c.481C>T | p.(Arg161*) | Homozygous | 36 | 5/282860 | P | [22] |
| #21 | GNAT2 | c.730_743del c.481C>T | p.(His244Serfs*7) p.(Arg161*) | Heterozygous | 34 36 | 5/152144 | LP (PVS1, PM2) P | Novel [22] |
| Characteristics | CNGA3 | CNGB3 | PDE6C | GNAT2 | p-Value (PDE6C vs. CNGA3) |
|---|---|---|---|---|---|
| n, (%) | 8 (38.1) | 3 (14.3) | 8 (38.1) | 2 (9.5) | N/A |
| Age | 7.6 ± 10.2 (0.6–30.7) | 13.8 ± 21.2 (0.5–38.2) | 13.2 ± 15.0 (0.6–47.2) | 10.8 ± 1.6 (9.7–11.9) | 0.430 a |
| BCVA (LogMAR) | |||||
| Right | 0.93 ± 0.35 (0.52–1.40) | 1.12 ± 0.26 (0.82–1.30) | 1.00 ± 0.16 (0.82–1.30) | 1.26 ± 0.37 (1.00–1.52) | 0.443 a |
| Left | 0.95 ± 0.29 (0.52–1.40) | 1.10 ± 0.17 (1.00–1.30) | 1.01 ± 0.21 (0.82–1.30) | 1.26 ± 0.37 (1.00–1.52) | 0.676 a |
| Fundus | 0.535 b | ||||
| Normal | 6 | 3 | 5 | 2 | |
| RPE change | 1 | 0 | 0 | 0 | |
| Retina/RPE atrophy | 0 | 0 | 2 | 0 | |
| ERG | |||||
| Rod response | Normal/subnormal | ||||
| Cone response | Absent | ||||
| Foveal hypoplasia | 0.386 b | ||||
| Yes | 3 | 2 | 1 | 0 | |
| No | 5 | 0 | 6 | 2 | |
| OCT grade (worse eye) | 0.011 (1 vs. 2,3,4) b | ||||
| 1 | 5 | 0 | 0 | 2 | |
| 2 | 3 | 0 | 3 | 0 | |
| 3 | 0 | 2 | 3 | 0 | |
| 4 | 0 | 0 | 2 | 0 | |
| Nation | Number of Patients, n | Mutated Genes, (%) | Remarks | Reference | |||
|---|---|---|---|---|---|---|---|
| CNGA3 | CNGB3 | PDE6C | GNAT2 | ||||
| Korea | 19 | 38.1 | 14.3 | 38.1 | 9.5 | Longitudinal analysis | This study |
| China | 119 | 81.5 | 5.9 | 10.1 | 0.8 | Identification of variants in ATF6 (1.7%) | [7] |
| Netherland | 63 | 4.8 | 87 | N/A | 0 | High proportion of CNGB3:p.T383IfsX13 (80%) | [35] |
| UK/US | 40 | 45 | 37.5 | 2.5 | 10 | Long term follow-up | [1] |
| Italia | 18 | 38.9 | 27.8 | 5.6 | 16.7 | High proportion of homozygotes (61.9%) | [4] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, Y.J.; Joo, K.; Lim, H.T.; Kim, S.S.; Han, J.; Woo, S.J. Clinical and Genetic Features of Korean Patients with Achromatopsia. Genes 2023, 14, 519. https://doi.org/10.3390/genes14020519
Choi YJ, Joo K, Lim HT, Kim SS, Han J, Woo SJ. Clinical and Genetic Features of Korean Patients with Achromatopsia. Genes. 2023; 14(2):519. https://doi.org/10.3390/genes14020519
Chicago/Turabian StyleChoi, Yong Je, Kwangsic Joo, Hyun Taek Lim, Sung Soo Kim, Jinu Han, and Se Joon Woo. 2023. "Clinical and Genetic Features of Korean Patients with Achromatopsia" Genes 14, no. 2: 519. https://doi.org/10.3390/genes14020519
APA StyleChoi, Y. J., Joo, K., Lim, H. T., Kim, S. S., Han, J., & Woo, S. J. (2023). Clinical and Genetic Features of Korean Patients with Achromatopsia. Genes, 14(2), 519. https://doi.org/10.3390/genes14020519