
Short Insertion and Deletion Discoveries via Whole-Genome Sequencing of 101 Thoroughbred Racehorses
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Whole-Genome Sequencing Data from 101 Thoroughbred Horses
2.2. INDEL Calling and Filtering
2.3. Statistical Analyses of Identified INDELs
3. Results
3.1. Numbers of Detected INDELs
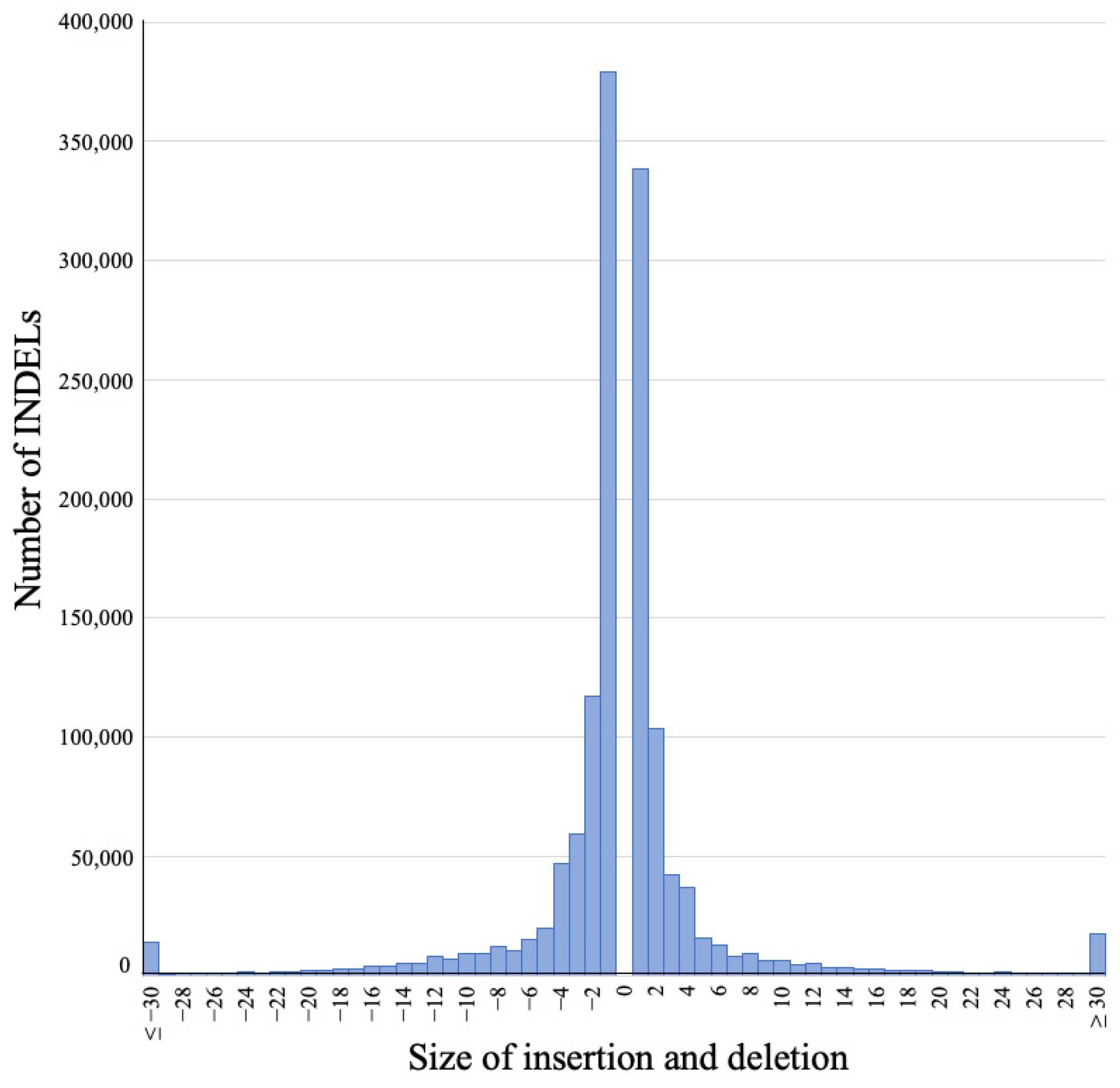
3.2. Sizes of Detected INDELs
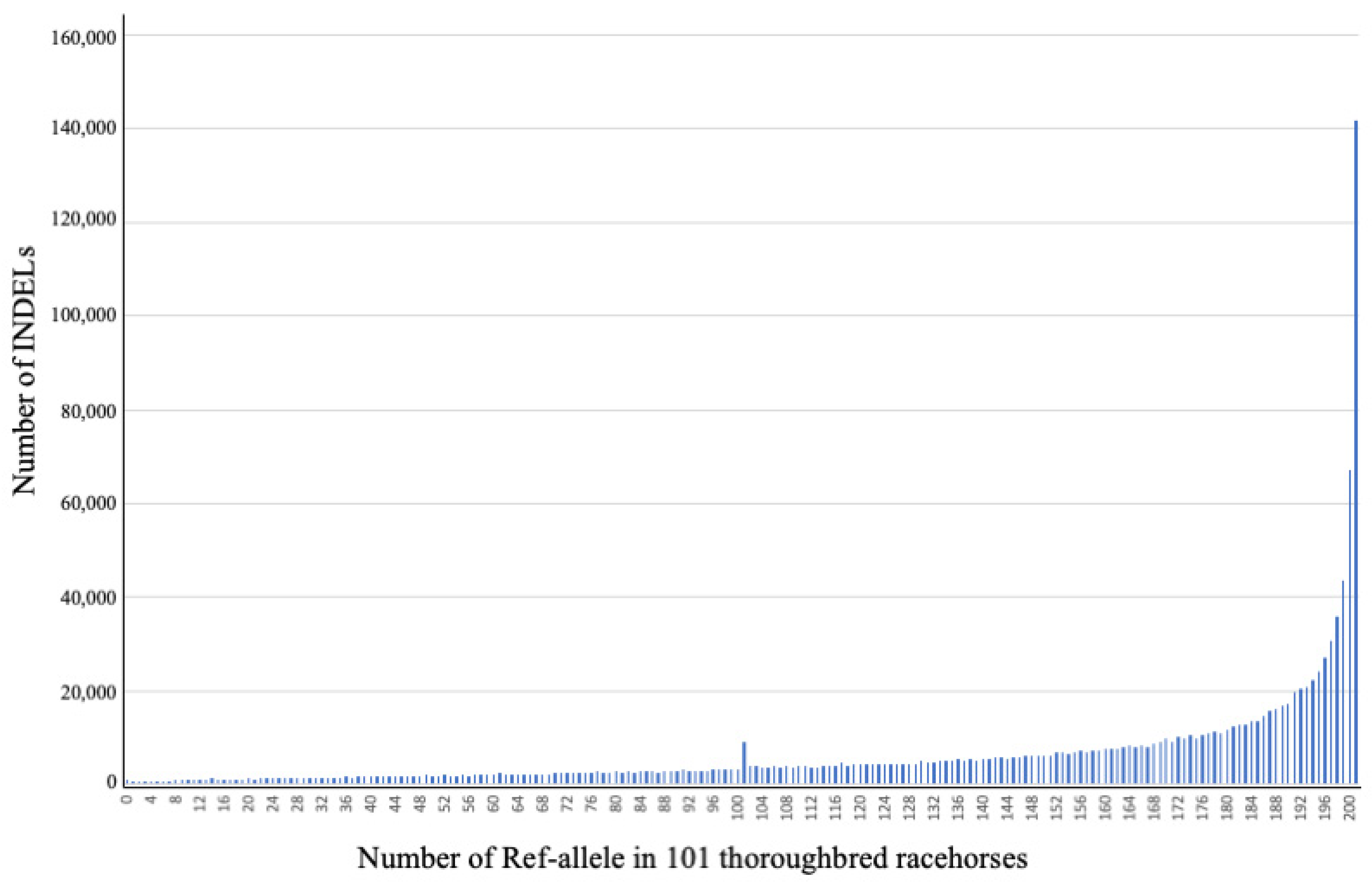
3.3. Allelic Frequency Distribution of INDELs Detected
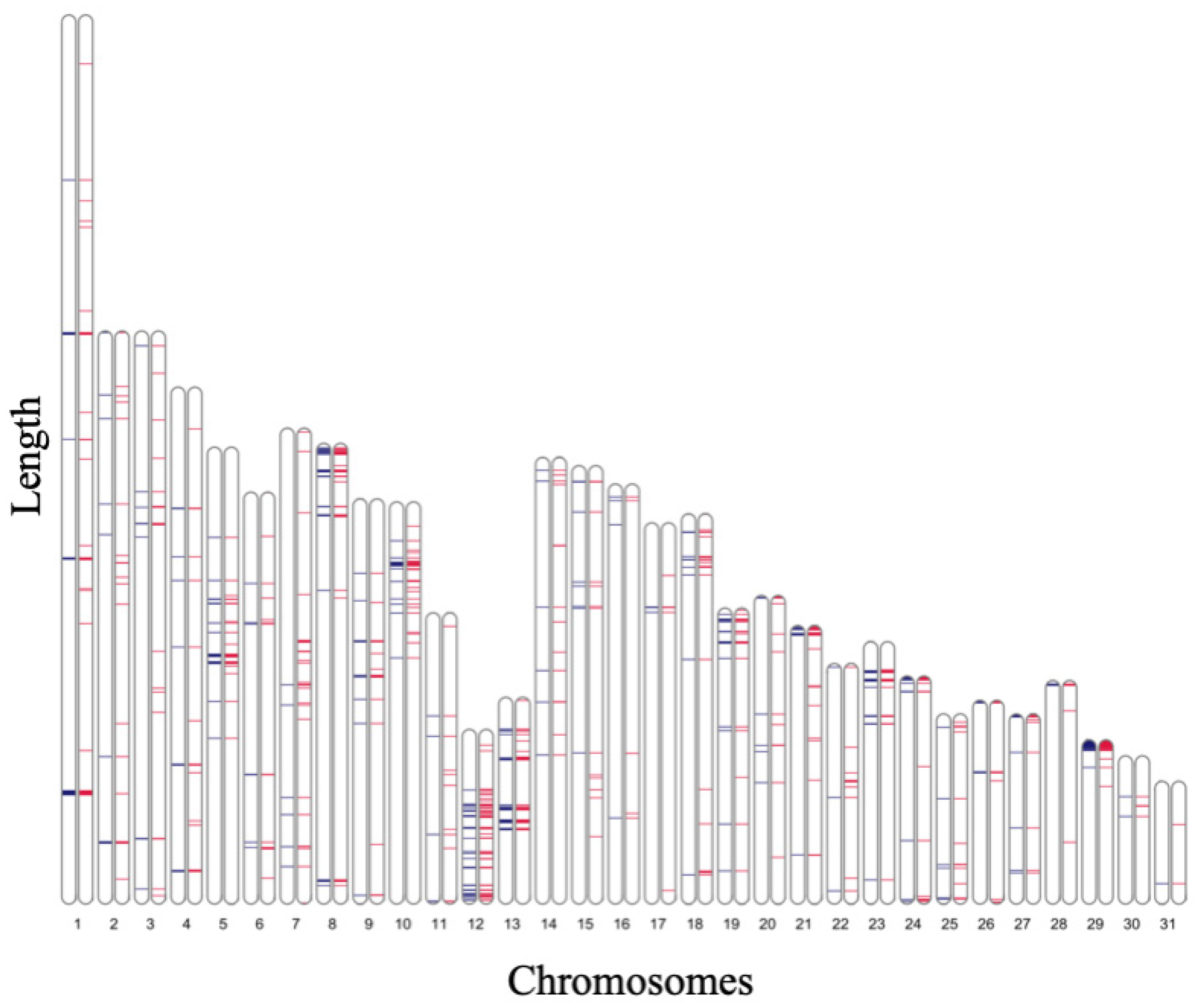
3.4. Detection of Genomic Regions Having MAF of 0.5 and All Heterozygous
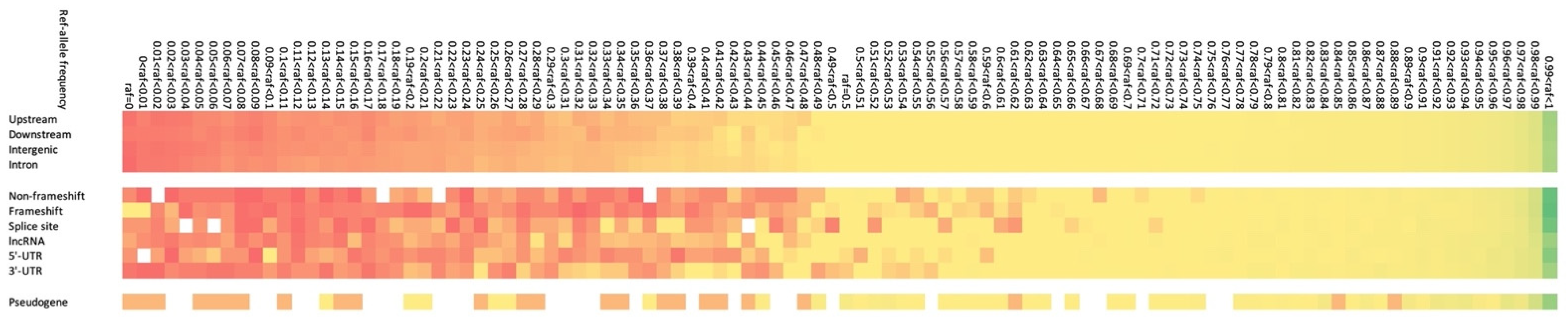
3.5. Characterisation of INDELs Detected
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bower, M.A.; Campana, M.G.; Whitten, M.; Edwards, C.J.; Jones, H.; Barrett, E.; Cassidy, R.; Nisbet, R.E.; Hill, E.W.; Howe, C.J.; et al. The cosmopolitan maternal heritage of the Thoroughbred racehorse breed shows a significant contribution from British and Irish native mares. Biol. Lett. 2011, 7, 316–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Stud Book Committee (Resouces/Thoroughbred Horse Statistics). Available online: https://www.internationalstudbook.com/resources/ (accessed on 2 February 2023).
- Wade, C.M.; Giulotto, E.; Sigurdsson, S.; Zoli, M.; Gnerre, S.; Imsland, F.; Lear, T.L.; Adelson, D.L.; Bailey, E.; Bellone, R.R.; et al. Genome sequence, comparative analysis, and population genetics of the domestic horse. Science 2009, 326, 865–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalbfleisch, T.S.; Rice, E.S.; DePriest, M.S., Jr.; Walenz, B.P.; Hestand, M.S.; Vermeesch, J.R.; O’Connell, B.L.; Fiddes, I.T.; Vershinina, A.O.; Saremi, N.F.; et al. Improved reference genome for the domestic horse increases assembly contiguity and composition. Commun. Biol. 2018, 1, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagannathan, V.; Gerber, V.; Rieder, S.; Tetens, J.; Thaller, G.; Drögemüller, C.; Leeb, T. Comprehensive characterization of horse genome variation by whole-genome sequencing of 88 horses. Anim. Genet. 2019, 50, 74–77. [Google Scholar] [CrossRef]
- Durward-Akhurst, S.A.; Schaefer, R.J.; Grantham, B.; Carey, W.K.; Mickelson, J.R.; McCue, M.E. Genetic variation and the distribution of variant types in the horse. Front. Genet. 2022, 12, 758366. [Google Scholar] [CrossRef]
- Tozaki, T.; Ohnuma, A.; Kikuchi, M.; Ishige, T.; Kakoi, H.; Hirota, K.; Kusano, K.; Nagata, S. Rare and common variant discovery by whole-genome sequencing of 101 Thoroughbred racehorses. Sci. Rep. 2021, 11, 16057. [Google Scholar] [CrossRef]
- Tozaki, T.; Hamilton, N.A. Control of gene doping in human and horse sports. Gene Ther. 2022, 29, 107–112. [Google Scholar] [CrossRef]
- Moro, L.N.; Viale, D.L.; Bastón, J.I.; Arnold, V.; Suvá, M.; Wiedenmann, E.; Olguín, M.; Miriuka, S.; Vichera, G. Generation of myostatin edited horse embryos using CRISPR/Cas9 technology and somatic cell nuclear transfer. Sci. Rep. 2020, 10, 15587. [Google Scholar] [CrossRef]
- Kim, D.E.; Lee, J.H.; Ji, K.B.; Park, K.S.; Kil, T.Y.; Koo, O.; Kim, M.K. Generation of genome-edited dogs by somatic cell nuclear transfer. BMC Biotechnol. 2022, 22, 19. [Google Scholar] [CrossRef]
- Sheets, T.P.; Park, C.H.; Park, K.E.; Powell, A.; Donovan, D.M.; Telugu, B.P. Somatic Cell Nuclear Transfer Followed by CRIPSR/Cas9 Microinjection Results in Highly Efficient Genome Editing in Cloned Pigs. Int. J. Mol. Sci. 2016, 17, 2031. [Google Scholar] [CrossRef] [Green Version]
- Hill, E.W.; Gu, J.; Eivers, S.S.; Fonseca, R.G.; McGivney, B.A.; Govindarajan, P.; Orr, N.; Katz, L.M.; MacHugh, D.E. A sequence polymorphism in MSTN predicts sprinting ability and racing stamina in thoroughbred horses. PLoS ONE 2010, 5, e8645. [Google Scholar] [CrossRef]
- Tozaki, T.; Sato, F.; Hill, E.W.; Miyake, T.; Endo, Y.; Kakoi, H.; Gawahara, H.; Hirota, K.; Nakano, Y.; Nambo, Y.; et al. Sequence variants at the myostatin gene locus influence the body composition of Thoroughbred horses. J. Vet. Med. Sci. 2011, 73, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Tozaki, T.; Ohnuma, A.; Nakamura, K.; Hano, K.; Takasu, M.; Takahashi, Y.; Tamura, N.; Sato, F.; Shimizu, K.; Kikuchi, M.; et al. Detection of indiscriminate genetic manipulation in Thoroughbred racehorses by targeted resequencing for gene-doping control. Genes 2022, 13, 1589. [Google Scholar] [CrossRef]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tozaki, T.; Ohnuma, A.; Kikuchi, M.; Ishige, T.; Kakoi, H.; Hirota, K.; Kusano, K.; Nagata, S. Identification of processed pseudogenes in the genome of Thoroughbred horses: Possibility of gene-doping detection considering the presence of pseudogenes. Anim. Genet. 2022, 53, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, W.P.; Francioli, L.C.; Hormozdiari, F.; Marschall, T.; Hehir-Kwa, J.Y.; Abdellaoui, A.; Lameijer, E.W.; Moed, M.H.; Koval, V.; Renkens, I.; et al. Characteristics of de novo structural changes in the human genome. Genome Res. 2015, 25, 792–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Abri, M.A.; Holl, H.M.; Kalla, S.E. Sutter NB, Brooks SA. Whole genome detection of sequence and structural polymorphism in six diverse horses. PLoS ONE 2020, 15, e0230899. [Google Scholar] [CrossRef] [Green Version]
- Miller, D.; Tallmadge, R.L.; Binns, M.; Zhu, B.; Mohamoud, Y.A.; Ahmed, A.; Brooks, S.A.; Antczak, D.F. Polymorphism at expressed DQ and DR loci in five common equine MHC haplotypes. Immunogenetics 2017, 69, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Tallmadge, R.L.; Lear, T.L.; Antczak, D.F. Genomic characterization of MHC class I genes of the horse. Immunogenetics 2005, 57, 763–774. [Google Scholar] [CrossRef]
- Rieder, S.; Taourit, S.; Mariat, D.; Langlois, B.; Guérin, G. Mutations in the agouti (ASIP), the extension (MC1R), and the brown (TYRP1) loci and their association to coat color phenotypes in horses (Equus caballus). Mamm. Genome 2001, 12, 450–455. [Google Scholar] [CrossRef]
- Kakoi, H.; Tozaki, T.; Nagata, S.; Gawahara, H.; Kijima-Suda, I. Development of a method for simultaneously genotyping multiple horse coat colour loci and genetic investigation of basic colour variation in Thoroughbred and Misaki horses in Japan. J. Anim. Breed. Genet. 2009, 126, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.F.; Hill, E.W.; Kelly, V.P.; Porter, R.K. The “speed gene” effect of myostatin arises in Thoroughbred horses due to a promoter proximal SINE insertion. PLoS ONE 2018, 13, e0205664. [Google Scholar] [CrossRef] [Green Version]
- Petersen, J.L.; Valberg, S.J.; Mickelson, J.R.; McCue, M.E. Haplotype diversity in the equine myostatin gene with focus on variants associated with race distance propensity and muscle fiber type proportions. Anim. Genet. 2014, 45, 827–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Guo, J.T. Structural and functional analysis of somatic coding and UTR indels in breast and lung cancer genomes. Sci. Rep. 2021, 11, 21178. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Lang, M.; Li, L.; Luo, H.; Hou, Y. Forensic features and genetic background exploration of a new 47-autosomal InDel panel in five representative Han populations residing in Northern China. Mol. Genet. Genom. Med. 2020, 8, e1224. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Nie, S.; Hu, L.; Fang, Y.; Cui, W.; Xu, H.; Zhao, C.; Zhu, B.F. Forensic efficacy evaluation and genetic structure exploration of the Yunnan Miao group by a multiplex InDel panel. Electrophoresis 2022, 43, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, C.; Xiao, C.; Chen, X.; Han, X.; Yi, S.; Huang, D. Mutation analysis of 28 autosomal short tandem repeats in the Chinese Han population. Mol. Biol. Rep. 2021, 48, 5363–5369. [Google Scholar] [CrossRef] [PubMed]
- Kakoi, H.; Nagata, S.; Kurosawa, M. DNA Typing with 17 microsatellites for parentage verification of racehorses in Japan. Anim. Sci. J. 2001, 72, 453–460. [Google Scholar] [CrossRef]
- Tozaki, T.; Kakoi, H.; Mashima, S.; Hirota, K.; Hasegawa, T.; Ishida, N.; Miura, N.; Choi-Miura, N.H.; Tomita, M. Population study and validation of paternity testing for Thoroughbred horses by 15 microsatellite loci. J. Vet. Med. Sci. 2001, 63, 1191–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, K.; Kakoi, H.; Gawahara, H.; Hasegawa, T.; Tozaki, T. Construction and validation of parentage testing for thoroughbred horses by 53 single nucleotide polymorphisms. J. Vet. Med. Sci. 2010, 72, 719–726. [Google Scholar] [CrossRef] [Green Version]
- Holl, H.M.; Vanhnasy, J.; Everts, R.E.; Hoefs-Martin, K.; Cook, D.; Brooks, S.A.; Carpenter, M.L.; Bustamante, C.D.; Lafayette, C. Single nucleotide polymorphisms for DNA typing in the domestic horse. Anim. Genet. 2017, 48, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Tozaki, T.; Ohnuma, A.; Kikuchi, M.; Ishige, T.; Kakoi, H.; Hirota, K.; Kusano, K.; Nagata, S. Microfluidic quantitative PCR detection of 12 transgenes from horse plasma for gene doping control. Genes 2020, 11, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tozaki, T.; Kwak, H.G.; Nakamura, K.; Takasu, M.; Ishii, H.; Ohnuma, A.; Kikuchi, M.; Ishige, T.; Kakoi, H.; Hirota, K.; et al. Sequence determination of phosphorothioated oligonucleotides using MALDI-TOF mass spectrometry for controlling gene doping in equestrian sports. Drug Test. Anal. 2022, 14, 175–180. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| INDEL Category | Chromosomes 1 to 31 | Chromosome X | Mitochondria |
|---|---|---|---|
| All INDELs | 1,453,349 | 113,047 | 18 |
| Diallelic INDELs | 1,274,708 | 101,966 | 16 |
| Multiallelic INDELs | 178,641 | 11,081 | 2 |
| Region | Chromosomes 1 to 31 | Chromosome X | Mitochondria |
|---|---|---|---|
| Upstream | 101,434 | 5633 | 16 |
| 5′-UTR | 2071 | 85 | 0 |
| Exon | 12,432 | 870 | 7 |
| Intron | 508,310 | 33,247 | 0 |
| 3′-UTR | 3258 | 227 | 0 |
| Downstream | 96,718 | 5989 | 16 |
| Intergenic | 754,952 | 67,934 | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tozaki, T.; Ohnuma, A.; Kikuchi, M.; Ishige, T.; Kakoi, H.; Hirota, K.-i.; Takahashi, Y.; Nagata, S.-i. Short Insertion and Deletion Discoveries via Whole-Genome Sequencing of 101 Thoroughbred Racehorses. Genes 2023, 14, 638. https://doi.org/10.3390/genes14030638
Tozaki T, Ohnuma A, Kikuchi M, Ishige T, Kakoi H, Hirota K-i, Takahashi Y, Nagata S-i. Short Insertion and Deletion Discoveries via Whole-Genome Sequencing of 101 Thoroughbred Racehorses. Genes. 2023; 14(3):638. https://doi.org/10.3390/genes14030638
Chicago/Turabian StyleTozaki, Teruaki, Aoi Ohnuma, Mio Kikuchi, Taichiro Ishige, Hironaga Kakoi, Kei-ichi Hirota, Yuji Takahashi, and Shun-ichi Nagata. 2023. "Short Insertion and Deletion Discoveries via Whole-Genome Sequencing of 101 Thoroughbred Racehorses" Genes 14, no. 3: 638. https://doi.org/10.3390/genes14030638