

Identification of Selection Signatures and Loci Associated with Important Economic Traits in Yunan Black and Huainan Pigs
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Approval
2.2. DNA Sample Collection
2.3. Single-Nucleotide Polymorphism (SNP) Genotyping and Data Quality Control
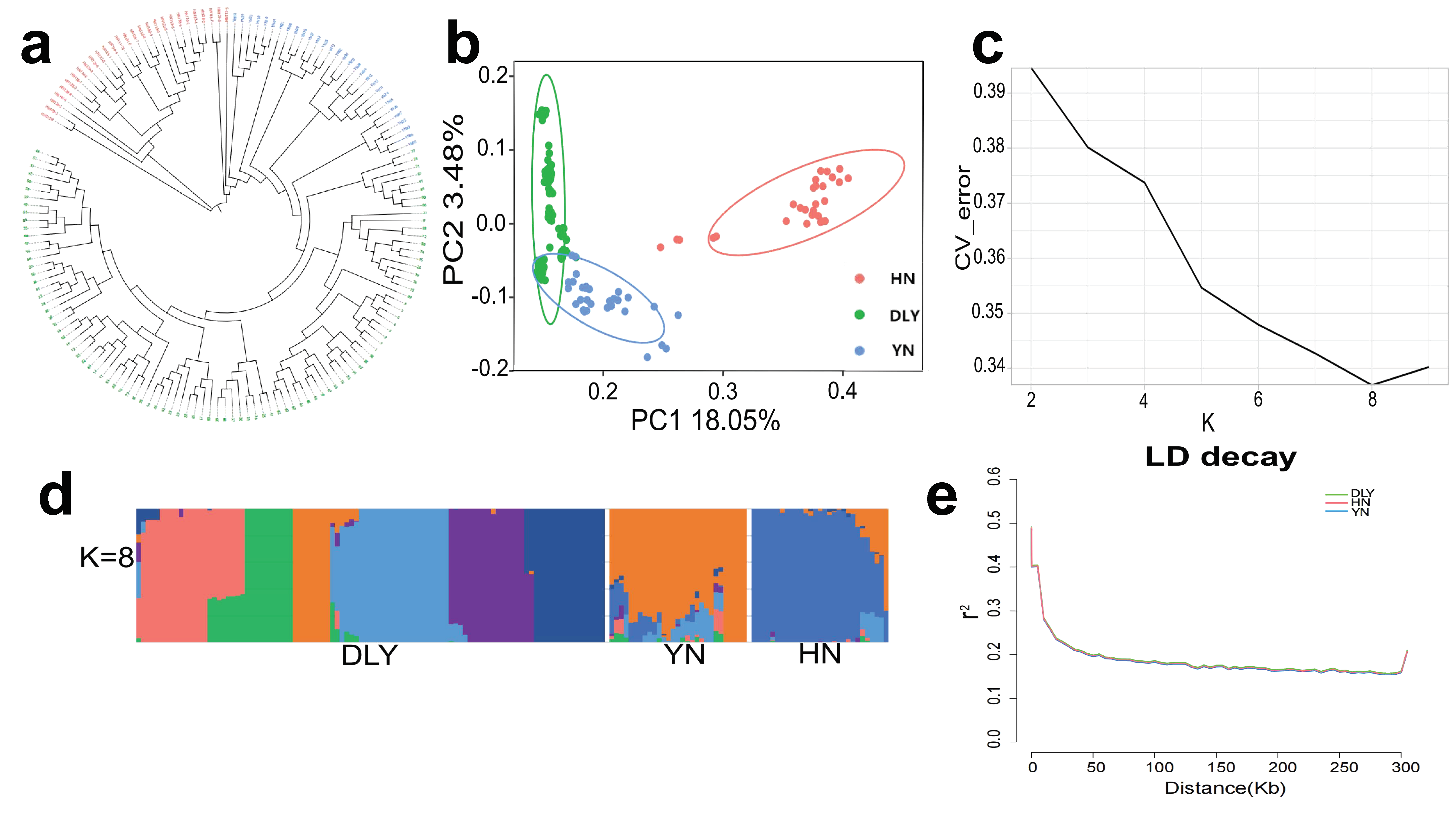
2.4. Diversity, Phylogenetic, and Population Genetic Analyses
2.5. Genome-Wide Selection Sweeps Detection
2.6. Annotation of Promoters of Tissue-Specific Expression Genes in 14 Tissues
2.7. Annotation of the Candidate Genes and Quantitative Trait Loci (QTL) Overlapping with Potential Selection Signatures
3. Results
3.1. Genotypes and Population Genetics Analysis
3.2. Identification of Signatures of Selection within-Population by CLR
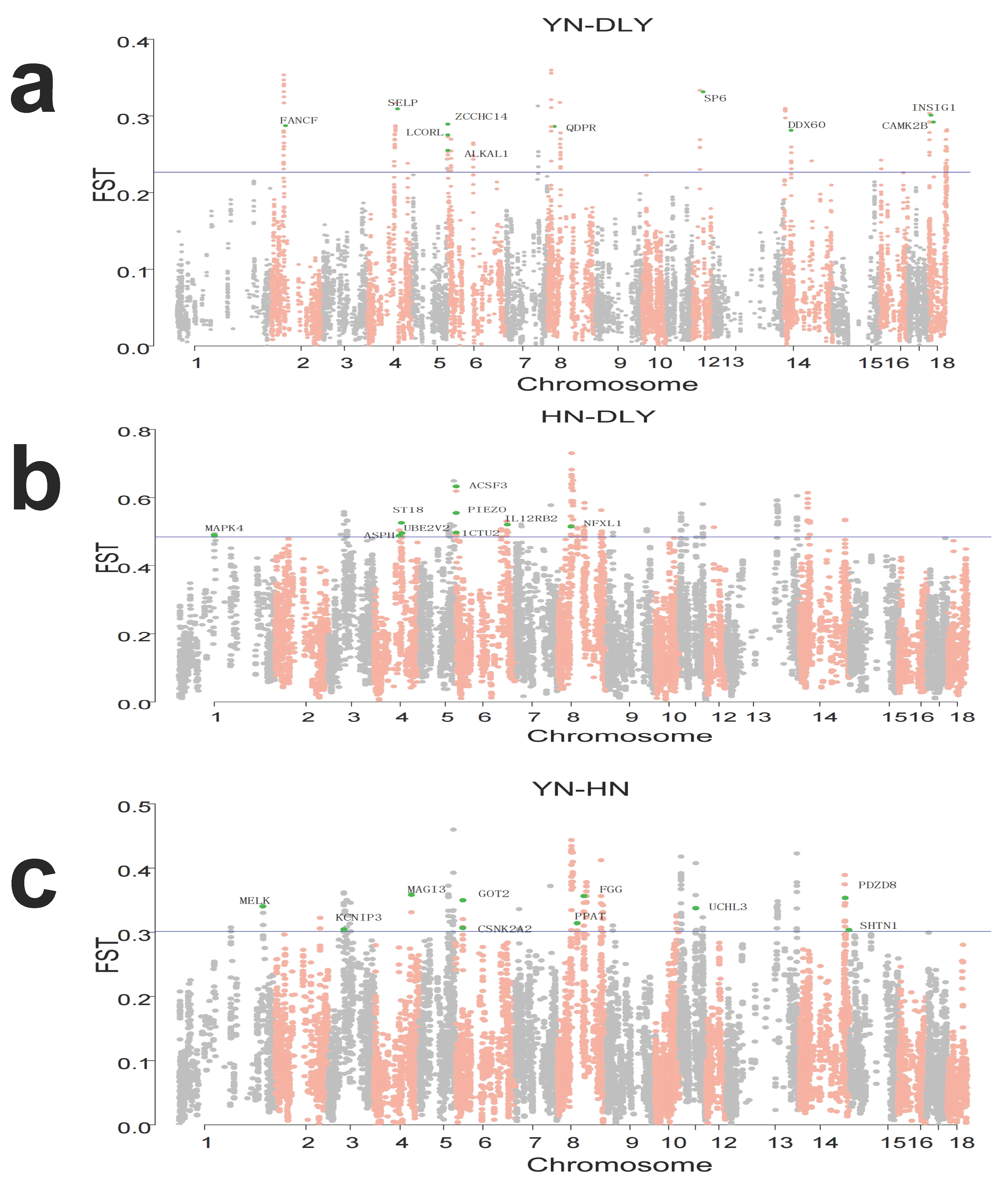
3.3. Identification of Signatures of Selection between-Population by Fst
3.4. QTL Overlapping with Potential Signatures of Selection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Briggs, H.M. Modern Breeds of Livestock Hardcover; Collier Macmillan Ltd.: New York, NY, USA, 1969; pp. 293–379. ISBN 978-002-314-720-3. [Google Scholar]
- China National Commission of Animal Genetic Resources. In Animal Genetic Resources in China: Pigs; China Agriculture Press: Beijing, China, 2011; pp. 60–63, 441–444. ISBN 978-710-915-882-5.
- Wang, K.; Wu, P.; Yang, Q.; Chen, D.; Zhou, J.; Jiang, A.; Ma, J.; Tang, Q.; Xiao, W.; Jiang, Y.; et al. Detection of Selection Signatures in Chinese Landrace and Yorkshire Pigs Based on Genotyping-by-Sequencing Data. Front. Genet. 2018, 9, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Zhang, H.; Zhang, Q.; Ding, X. Identification of selection footprints on the X chromosome in pig. PLoS ONE 2014, 9, e94911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Ruan, Y.; Sun, J.; Shi, P.; Huang, J.; Dai, L.; Xiao, M.; Xu, H. Association analysis of PRKAA2 and MSMB polymorphisms and growth traits of Xiangsu hybrid pigs. Genes 2022, 14, 113. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, C.; Meng, F.; Yao, Z.; Fan, Z.; Yang, Y.; Meng, X.; Zhan, Y.; Sun, Y.; Ma, F.; et al. Genome-wide association studies for flesh color and intramuscular fat in (Duroc x landrace x large white) crossbred commercial pigs. Genes 2022, 13, 2131. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, X.; Li, M.; Sun, H.; Chen, Q.; Yan, D.; Dong, X.; Pan, Y.; Lu, S. Genome-wide association study of growth traits in a four-way crossbred pig population. Genes 2022, 13, 1990. [Google Scholar] [CrossRef]
- Li, M.; Tian, S.; Jin, L.; Zhou, G.; Li, Y.; Zhang, Y.; Wang, T.; Yeung, C.K.; Chen, L.; Ma, J.; et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 2013, 45, 1431–1438. [Google Scholar] [CrossRef]
- Ai, H.; Fang, X.; Yang, B.; Huang, Z.; Chen, H.; Mao, L.; Zhang, F.; Zhang, L.; Cui, L.; He, W.; et al. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing. Nat. Genet. 2015, 47, 217–225. [Google Scholar] [CrossRef]
- Frantz, L.A.; Schraiber, J.G.; Madsen, O.; Megens, H.J.; Cagan, A.; Bosse, M.; Paudel, Y.; Crooijmans, R.P.; Larson, G.; Groenen, M.A. Evidence of long-term gene flow and selection during domestication from analyses of Eurasian wild and domestic pig genomes. Nat. Genet. 2015, 47, 1141–1148. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, S.; Zhang, K.; Fang, C.; Xie, S.; Du, X.; Li, X.; Ni, D.; Zhao, S. Genomic analysis to identify signatures of artificial selection and loci associated with important economic traits in duroc pigs. G3 (Bethesda) 2018, 8, 3617–3625. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wu, X.; Wang, J.; Hou, Y.; Liu, Y.; Wang, B.; Hu, X.; Zheng, X.; Zhang, X.; Ding, Y.; et al. Detection of selection signatures in Anqing six-end-white pigs based on resequencing data. Genes 2022, 13, 2310. [Google Scholar] [CrossRef]
- Guo, L.; Sun, H.; Zhao, Q.; Xu, Z.; Zhang, Z.; Liu, D.; Qadri, Q.R.; Ma, P.; Wang, Q.; Pan, Y. Positive selection signatures in Anqing six-end-white pig population based on reduced-representation genome sequencing data. Anim. Genet. 2021, 52, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Diao, S.; Huang, S.; Chen, Z.; Teng, J.; Ma, Y.; Yuan, X.; Chen, Z.; Zhang, H.; Li, J.; Zhang, Z. Genome-wide signatures of selection detection in three south china indigenous pigs. Genes 2019, 10, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Yu, Y.; Feng, W.; Du, H.; Yu, J.; Kang, H.; Zheng, X.; Wang, Z.; Liu, G.E.; Ernst, C.W.; et al. Evidence of evolutionary history and selective sweeps in the genome of Meishan pig reveals its genetic and phenotypic characterization. Gigascience 2018, 7, giy058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.; Feng, J.; Xu, Y.; Fang, Y.; Zhao, W.; Zhang, Z.; Zhang, Z.; Li, M.; Wang, Q.; Pan, Y. Genomic signatures reveal breeding effects of Lulai pigs. Genes 2022, 13, 1969. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Nielsen, R.; Williamson, S.; Kim, Y.; Hubisz, M.J.; Clark, A.G.; Bustamante, C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005, 15, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Yao, Y.; Yin, H.; Cai, Z.; Wang, Y.; Bai, L.; Kern, C.; Halstead, M.; Chanthavixay, G.; Trakooljul, N.; et al. Pig genome functional annotation enhances the biological interpretation of complex traits and human disease. Nat. Commun. 2021, 12, 5848. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Huang Da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.L.; Park, C.A.; Reecy, J.M. Building a livestock genetic and genomic information knowledgebase through integrative developments of Animal QTLdb and CorrDB. Nucleic Acids Res. 2019, 47, D701–D710. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the future: Meeting new challenges and providing updated services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar] [CrossRef]
- Chen, H.; Patterson, N.; Reich, D. Population differentiation as a test for selective sweeps. Genome Res. 2010, 20, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Gianola, D.; Simianer, H.; Qanbari, S. A two-step method for detecting selection signatures using genetic markers. Genet. Res. (Camb) 2010, 92, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006, 4, e72. [Google Scholar] [CrossRef] [Green Version]
- Han, G.S.; Carman, G.M. Characterization of the human LPIN1-encoded phosphatidate phosphatase isoforms. J. Biol. Chem. 2010, 285, 14628–14638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.Y.; Song, L.; Sun, Y.; Li, J.; Yi, C.; Lam, S.M.; Xu, D.; Zhou, L.; Li, X.; Yang, Y.; et al. Tip60-mediated lipin 1 acetylation and ER translocation determine triacylglycerol synthesis rate. Nat. Commun. 2018, 9, 1916. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Wang, J.; Liu, B.; Pan, W.; Farr, G.H., 3rd; Flynn, C.; Yuan, H.; Takada, S.; Kimelman, D.; Li, L.; et al. Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol. Cell 2001, 7, 801–809. [Google Scholar] [CrossRef]
- Sobrier, M.L.; Tsai, Y.C.; Pérez, C.; Leheup, B.; Bouceba, T.; Duquesnoy, P.; Copin, B.; Sizova, D.; Penzo, A.; Stanger, B.Z.; et al. Functional characterization of a human POU1F1 mutation associated with isolated growth hormone deficiency: A novel etiology for IGHD. Hum. Mol. Genet. 2016, 25, 472–483. [Google Scholar] [CrossRef] [Green Version]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.; Hong, D.; Iborra, F.; Sarno, S.; Enver, T. NOV (CCN3) functions as a regulator of human hematopoietic stem or progenitor cells. Science 2007, 316, 590–593. [Google Scholar] [CrossRef]
- Shimoyama, T.; Hiraoka, S.; Takemoto, M.; Koshizaka, M.; Tokuyama, H.; Tokuyama, T.; Watanabe, A.; Fujimoto, M.; Kawamura, H.; Sato, S.; et al. CCN3 inhibits neointimal hyperplasia through modulation of smooth muscle cell growth and migration. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Yamaguchi, S.; Ando, R.; Miyawaki, A.; Kabasawa, Y.; Takagi, M.; Li, C.L.; Perbal, B.; Katsube, K. The nephroblastoma overexpressed gene (NOV/ccn3) protein associates with Notch1 extracellular domain and inhibits myoblast differentiation via Notch signaling pathway. J. Biol. Chem. 2002, 277, 29399–29405. [Google Scholar] [CrossRef] [Green Version]
- Inoue, N.; Hess, K.D.; Moreadith, R.W.; Richardson, L.L.; Handel, M.A.; Watson, M.L.; Zinn, A.R. New gene family defined by MORC, a nuclear protein required for mouse spermatogenesis. Hum. Mol. Genet. 1999, 8, 1201–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.J.; Lee, Y.; Zhang, H.; Song, J.S.; Hu, J.C.; Simmer, J.P.; Kim, J.W. A Novel De Novo SP6 mutation causes severe hypoplastic amelogenesis imperfecta. Genes 2021, 12, 346. [Google Scholar] [CrossRef]
- N’Diaye, A.; Chen, G.K.; Palmer, C.D.; Ge, B.; Tayo, B.; Mathias, R.A.; Ding, J.; Nalls, M.A.; Adeyemo, A.; Adoue, V.; et al. Identification, replication, and fine-mapping of Loci associated with adult height in individuals of african ancestry. PLoS Genet. 2011, 7, e1002298. [Google Scholar] [CrossRef] [Green Version]
- Hirabayashi, Y.; Kwon, S.K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. ER-mitochondria tethering by PDZD8 regulates Ca(2+) dynamics in mammalian neurons. Science 2017, 358, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, I.; Paucar, A.A.; Bajpai, R.; Dougherty, J.D.; Zewail, A.; Kelly, T.K.; Kim, K.J.; Ou, J.; Groszer, M.; Imura, T.; et al. Maternal embryonic leucine zipper kinase (MELK) regulates multipotent neural progenitor proliferation. J. Cell Biol. 2005, 170, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Sloan, J.L.; Johnston, J.J.; Manoli, I.; Chandler, R.J.; Krause, C.; Carrillo-Carrasco, N.; Chandrasekaran, S.D.; Sysol, J.R.; O’Brien, K.; Hauser, N.S.; et al. Exome sequencing identifies ACSF3 as a cause of combined malonic and methylmalonic aciduria. Nat. Genet. 2011, 43, 883–886. [Google Scholar] [CrossRef] [Green Version]
- Witkowski, A.; Thweatt, J.; Smith, S. Mammalian ACSF3 protein is a malonyl-CoA synthetase that supplies the chain extender units for mitochondrial fatty acid synthesis. J. Biol. Chem. 2011, 286, 33729–33736. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Barghouth, M.; Dou, H.; Luan, C.; Wang, Y.; Karagiannopoulos, A.; Jiang, X.; Krus, U.; Fex, M.; Zhang, Q.; et al. A critical role of the mechanosensor PIEZO1 in glucose-induced insulin secretion in pancreatic β-cells. Nat. Commun. 2022, 13, 4237. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fst Top 1% Overlaps Candidate Regional Genes and Traits with Porcine QTL | ||||||
|---|---|---|---|---|---|---|
| Group | CHR | START | END | FST | QTL | Genes |
| YN_DLY | Chr.1 | 26,823,251 | 26,823,255 | 0.93663199 | Triglyceride level QTL (56,029) | DOLK, CRAT, PTPA, DOLPP1 |
| Chr.12 | 28,689,860 | 28,689,864 | 0.73554397 | Hemoglobin QTL (22,161) | PNPO | |
| Chr.12 | 29,033,566 | 29,033,570 | 0.61874989 | Loin muscle depth QTL (255,351) | NPEPPS, OSBPL7, SRCN2 | |
| Chr.14 | 136,511,430 | 136,511,434 | 0.93663199 | Number of mummified pigs QTL (178,885) | MYOF, CEP55, ADAM12, | |
| YN_HN | Chr.8 | 38,946,696 | 38,946,700 | 0.93663199 | Litter size QTL (130,289) | DCUN1D4, SPATA18, PAICS, PPAT |
| Chr.11 | 47,893,515 | 47,893,519 | 0.61874989 | Intramuscular fat content QTL (147,429) | UCHL3, KATNAL1 | |
| HN_DLY | Chr.1 | 100,049,958 | 141,109,887 | 0.93663199 | Ear weight QTL (8853) | MAPK4 |
| Chr.4 | 77,799,657 | 77,799,661 | 0.93663199 | Humerus length QTL (57,328) | UBE2V2 | |
| Chr.4 | 79,493,441 | 79,493,445 | 0.61874989 | Feed conversion ratio QTL (139,907) | ST18 | |
| Chr.6 | 145,288,072 | 145,288,076 | 0.73554397 | Linolenic acid content QTL (193,502) | PIEZO1, ACSF3, CTU2 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Y.; Tan, T.; Li, Z.; Ma, Z.; Lan, G.; Liang, J.; Li, K.; Bai, L. Identification of Selection Signatures and Loci Associated with Important Economic Traits in Yunan Black and Huainan Pigs. Genes 2023, 14, 655. https://doi.org/10.3390/genes14030655
Han Y, Tan T, Li Z, Ma Z, Lan G, Liang J, Li K, Bai L. Identification of Selection Signatures and Loci Associated with Important Economic Traits in Yunan Black and Huainan Pigs. Genes. 2023; 14(3):655. https://doi.org/10.3390/genes14030655
Chicago/Turabian StyleHan, Yachun, Tao Tan, Zixin Li, Zheng Ma, Ganqiu Lan, Jing Liang, Kui Li, and Lijing Bai. 2023. "Identification of Selection Signatures and Loci Associated with Important Economic Traits in Yunan Black and Huainan Pigs" Genes 14, no. 3: 655. https://doi.org/10.3390/genes14030655