
An in Silico Approach to Identifying TF Binding Sites: Analysis of the Regulatory Regions of BUSCO Genes from Fungal Species in the Ceratocystidaceae Family


Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Selection and Annotation for Motif Discovery
2.2. Motif Discovery
2.3. Motif Comparison
2.4. Novel Motif Identification in Non-Ceratocystidaceae Species
3. Results
3.1. Gene Selection for Motif Discovery
3.2. Motif Discovery in the Ceratocystidaceae Species
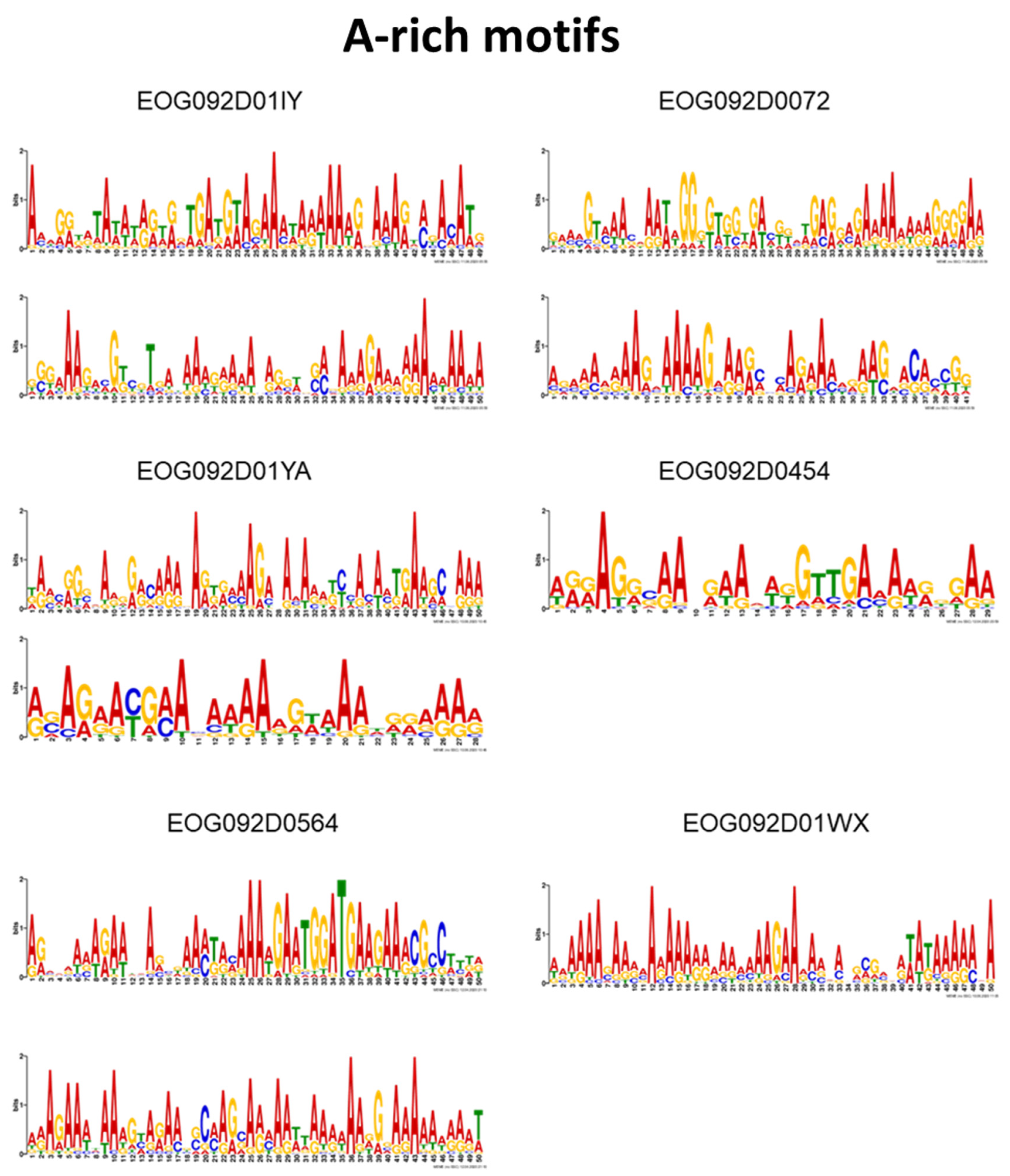
3.3. Motif Comparison to Known TF Binding Sites
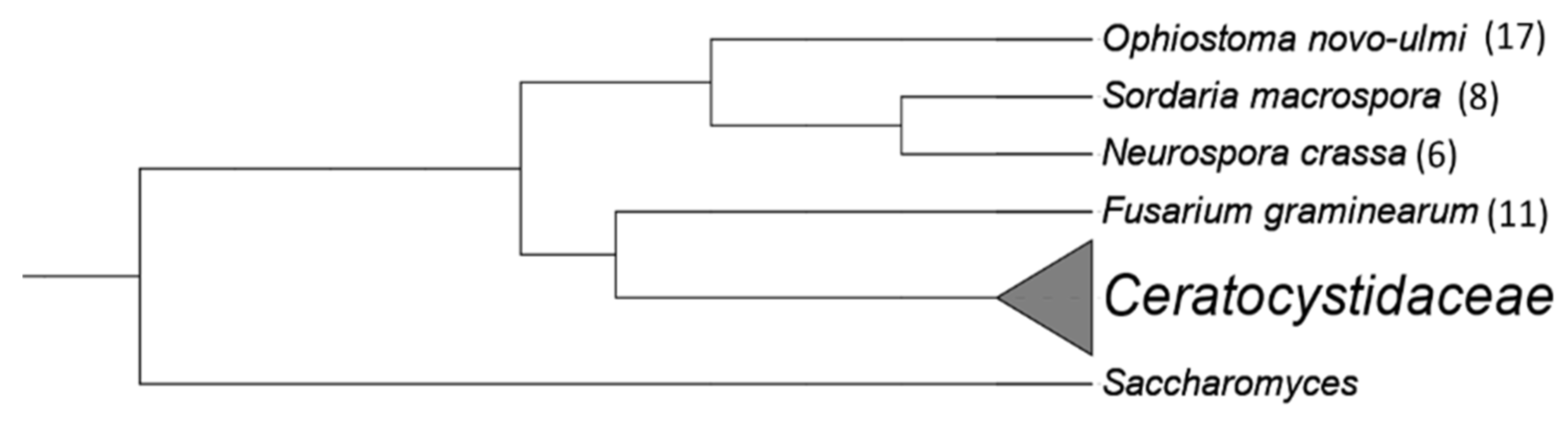
3.4. Identifying Novel Motifs in Non-Ceratocystidaceae
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Latchman, D.S. Transcription factors: An overview. Int. J. Biochem. Cell Biol. 1997, 29, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Mitsis, T.; Efthimiadou, A.; Bacopoulou, F.; Vlachakis, D.; Chrousos, G.P.; Eliopoulos, E. Transcription factors and evolution: An integral part of gene expression. World Acad. Sci. J. 2020, 2, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.-J.M.; Chou, L.-C.; Hsieh, T.-T.; Lee, D.-D.; Liu, K.-W.; Yu, C.-Y.; Oyang, Y.-J.; Tsai, H.-K.; Chen, C.-Y. De novo motif discovery facilitates identification of interactions between transcription factors in Saccharomyces cerevisiae. Bioinformatics 2012, 28, 701–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garner, M.M.; Revzin, A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: Application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res. 1981, 9, 3047–3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenowitz, M.; Senear, D.F.; Shea, M.A.; Ackers, G.K. Quantitative DNase footprint titration: A method for studying protein-DNA interactions. Methods Enzymol. 1986, 130, 132–181. [Google Scholar] [CrossRef]
- Yang, V.W. Eukaryotic transcription factors: Identification, characterization and functions. J. Nutr. 1998, 128, 2045–2051. [Google Scholar] [CrossRef] [Green Version]
- Zambelli, F.; Pesole, G.; Pavesi, G. Motif discovery and transcription factor binding sites before and after the next-generation sequencing era. Brief. Bioinform. 2013, 14, 225–237. [Google Scholar] [CrossRef]
- Hashim, F.A.; Mabrouk, M.S.; Al-Atabany, W. Review of different sequence motif finding algorithms. Avicenna J. Med. Biotechnol. 2019, 11, 130. [Google Scholar]
- Das, M.K.; Dai, H.-K. A survey of DNA motif finding algorithms. BMC Bioinf. 2007, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Simcha, D.; Price, N.D.; Geman, D. The limits of de novo DNA motif discovery. PLoS ONE 2012, 7, e47836. [Google Scholar] [CrossRef] [Green Version]
- Gasch, A.P.; Moses, A.M.; Chiang, D.Y.; Fraser, H.B.; Berardini, M.; Eisen, M.B. Conservation and evolution of cis-regulatory systems in ascomycete fungi. PLoS Biol. 2004, 2, e398. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Stormo, G.D. Combining phylogenetic data with co-regulated genes to identify regulatory motifs. Bioinformatics 2003, 19, 2369–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Helden, J.; André, B.; Collado-Vides, J. Extracting regulatory sites from the upstream region of yeast genes by computational analysis of oligonucleotide frequencies. J. Mol. Biol. 1998, 281, 827–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandve, G.K.; Drabløs, F. A survey of motif discovery methods in an integrated framework. Biol. Direct 2006, 1, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacIsaac, K.D.; Wang, T.; Gordon, D.B.; Gifford, D.K.; Stormo, G.D.; Fraenkel, E. An improved map of conserved regulatory sites for Saccharomyces cerevisiae. BMC Bioinf. 2006, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, L.; Breakspear, A.; Kistler, C.; Ma, L.-J.; Xie, X. Systematic discovery of regulatory motifs in Fusarium graminearum by comparing four Fusarium genomes. BMC Genom. 2010, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Roux, J.; Wingfield, M.J. Ceratocystis species: Emerging pathogens of non-native plantation Eucalyptus and Acacia species. South. For. 2009, 71, 115. [Google Scholar] [CrossRef]
- De Beer, Z.W.; Duong, T.A.; Barnes, I.; Wingfield, B.D.; Wingfield, M.J. Redefining Ceratocystis and allied genera. Stud. Mycol. 2014, 79, 187–219. [Google Scholar] [CrossRef] [Green Version]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [Green Version]
- Wilken, P.M.; Aylward, J.; Chand, R.; Grewe, F.; Lane, F.A.; Sinha, S.; Ametrano, C.; Distefano, I.; Divakar, P.K.; Duong, T.A.; et al. IMA Genome—F13: Draft genome sequences of Ambrosiella cleistominuta, Cercospora brassicicola, C. citrullina, Physcia stellaris, and Teratosphaeria pseudoeucalypti. IMA Fungus 2020, 11, 19. [Google Scholar] [CrossRef]
- Vanderpool, D.; Bracewell, R.R.; McCutcheon, J.P. Know your farmer: Ancient origins and multiple independent domestications of ambrosia beetle fungal cultivars. Mol. Ecol. 2017, 27, 2077–2094. [Google Scholar] [CrossRef] [PubMed]
- Nel, W.J.; Duong, T.A.; Wingfield, B.D.; Wingfield, M.J.; de Beer, Z.W. A new genus and species for the globally important, multihost root pathogen Thielaviopsis basicola. Plant Pathol. 2018, 67, 871–882. [Google Scholar] [CrossRef]
- Wingfield, B.D.; Ambler, J.M.; Coetzee, M.; De Beer, Z.W.; Duong, T.A.; Joubert, F.; Hammerbacher, A.; McTaggart, A.R.; Naidoo, K.; Nguyen, H.D.; et al. IMA Genome-F 6: Draft genome sequences of Armillaria fuscipes, Ceratocystiopsis minuta, Ceratocystis adiposa, Endoconidiophora laricicola, E. polonica and Penicillium freii DAOMC 242723. IMA Fungus 2016, 7, 217–227. [Google Scholar] [CrossRef] [PubMed]
- van der Nest, M.A.; Beirn, L.A.; Crouch, J.A.; Demers, J.E.; de Beer, Z.W.; De Vos, L.; Gordon, T.R.; Moncalvo, J.-M.; Naidoo, K.; Sanchez-Ramirez, S.; et al. IMA Genome-F 3: Draft genomes of Amanita jacksonii, Ceratocystis albifundus, Fusarium circinatum, Huntiella omanensis, Leptographium procerum, Rutstroemia sydowiana, and Sclerotinia echinophila. IMA Fungus 2014, 5, 472–485. [Google Scholar] [CrossRef] [Green Version]
- Wilken, P.M.; Steenkamp, E.T.; De Beer, Z.W.; Wingfield, M.J.; Wingfield, B.D. IMA Genome-F1: Draft nuclear genome sequence for the plant pathogen, Ceratocystis fimbriata. IMA Fungus 2013, 4, 357–358. [Google Scholar] [CrossRef]
- Wingfield, B.D.; Barnes, I.; De Beer, Z.W.; De Vos, L.; Duong, T.A.; Kanzi, A.M.; Naidoo, K.; Nguyen, H.D.; Santana, Q.C.; Sayari, M.; et al. IMA Genome-F 5: Draft genome sequences of Ceratocystis eucalypticola, Chrysoporthe cubensis, C. deuterocubensis, Davidsoniella virescens, Fusarium temperatum, Graphilbum fragrans, Penicillium nordicum, and Thielaviopsis musarum. IMA Fungus 2015, 6, 493–506. [Google Scholar] [CrossRef]
- Wingfield, B.D.; Duong, T.A.; Hammerbacher, A.; van der Nest, M.A.; Wilson, A.; Chang, R.; de Beer, Z.W.; Steenkamp, E.T.; Wilken, P.M.; Naidoo, K.; et al. IMA Genome-F 7: Draft genome sequences for Ceratocystis fagacearum, C. harringtonii, Grosmannia penicillata, and Huntiella bhutanensis. IMA Fungus 2016, 7, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Van der Nest, M.A.; Bihon, W.; De Vos, L.; Naidoo, K.; Roodt, D.; Rubagotti, E.; Slippers, B.; Steenkamp, E.T.; Wilken, P.M.; Wilson, A.; et al. IMA Genome-F 2: Draft genome sequences of Diplodia sapinea, Ceratocystis manginecans, and Ceratocystis moniliformis. IMA Fungus 2014, 5, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Wingfield, B.D.; Bills, G.F.; Dong, Y.; Huang, W.; Nel, W.J.; Swalarsk-Parry, B.S.; Vaghefi, N.; Wilken, P.M.; An, Z.; de Beer, Z.W.; et al. IMA Genome-F 9: Draft genome sequence of Annulohypoxylon stygium, Aspergillus mulundensis, Berkeleyomyces basicola (syn. Thielaviopsis basicola), Ceratocystis smalleyi, two Cercospora beticola strains, Coleophoma cylindrospora, Fusarium fracticaudum, Phialophora cf. hyalina, and Morchella septimelata. IMA Fungus 2018, 9, 199–223. [Google Scholar] [CrossRef] [Green Version]
- Sayari, M.; van der Nest, M.A.; Steenkamp, E.T.; Soal, N.C.; Wilken, P.M.; Wingfield, B.D. Distribution and evolution of nonribosomal peptide synthetase gene clusters in the Ceratocystidaceae. Genes 2019, 10, 328. [Google Scholar] [CrossRef] [Green Version]
- Wingfield, B.D.; Liu, M.; Nguyen, H.D.; Lane, F.A.; Morgan, S.W.; De Vos, L.; Wilken, P.M.; Duong, T.A.; Aylward, J.; Coetzee, M.; et al. IMA Genome-F 10: Nine draft genome sequences of Claviceps purpurea s. lat., including C. arundinis, C. humidiphila, and C. cf. spartinae, pseudomolecules for the pitch canker pathogen Fusarium circinatum, draft genome of Davidsoniella eucalypti, Grosmannia galeiformis, Quambalaria eucalypti, and Teratosphaeria destructans. IMA Fungus 2018, 9, 401–418. [Google Scholar] [CrossRef] [PubMed]
- Wingfield, B.D.; Berger, D.K.; Steenkamp, E.T.; Lim, H.J.; Duong, T.A.; Bluhm, B.H.; de Beer, Z.W.; De Vos, L.; Fourie, G.; Naidoo, K.; et al. IMA Genome-F 8: Draft genome of Cercospora zeina, Fusarium pininemorale, Hawksworthiomyces lignivorus, Huntiella decipiens and Ophiostoma ips. IMA Fungus 2017, 8, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Van der Nest, M.A.; Steenkamp, E.T.; McTaggart, A.R.; Trollip, C.; Godlonton, T.; Sauerman, E.; Roodt, D.; Naidoo, K.; Coetzee, M.P.A.; Wilken, P.M.; et al. Saprophytic and pathogenic fungi in the Ceratocystidaceae differ in their ability to metabolize plant-derived sucrose. BMC Evol. Biol. 2015, 15, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilken, P.M.; Steenkamp, E.T.; Van der Nest, M.A.; Wingfield, M.J.; De Beer, Z.W.; Wingfield, B.D. Unexpected placement of the MAT1-1-2 gene in the MAT1-2 idiomorph of Thielaviopsis. Fungal Genet. Biol. 2018, 113, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Galagan, J.E.; Calvo, S.E.; Borkovich, K.A.; Selker, E.U.; Read, N.D.; Jaffe, D.; FitzHugh, W.; Ma, L.-J.; Smirnov, S.; Purcell, S.; et al. The genome sequence of the filamentous fungus Neurospora crassa. Nature 2003, 422, 859–868. [Google Scholar] [CrossRef] [Green Version]
- Nowrousian, M.; Stajich, J.E.; Chu, M.; Engh, I.; Espagne, E.; Halliday, K.; Kamerewerd, J.; Kempken, F.; Knab, B.; Kuo, H.-C.; et al. De novo assembly of a 40 Mb eukaryotic genome from short sequence reads: Sordaria macrospora, a model organism for fungal morphogenesis. PLoS Genet. 2010, 6, e1000891. [Google Scholar] [CrossRef] [Green Version]
- Wingfield, B.D.; Steenkamp, E.T.; Santana, Q.C.; Coetzee, M.P.A.; Bam, S.; Barnes, I.; Beukes, C.W.; Chan, W.Y.; de Vos, L.; Fourie, G.; et al. First fungal genome sequence from Africa: A preliminary analysis. S. Afr. J. Sci. 2012, 108, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Forgetta, V.; Leveque, G.; Dias, J.; Grove, D.; Lyons, R., Jr.; Genik, S.; Wright, C.; Singh, S.; Peterson, N.; Zianni, M.; et al. Sequencing of the Dutch elm disease fungus genome using the Roche/454 GS-FLX Titanium System in a comparison of multiple genomics core facilities. J. Biomol. Tech. 2013, 24, 39. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Bailey, T.; Boden, M.; Buske, F.; Frith, M.; Grant, C.; Clementi, L.; Ren, J.L. MEME suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, 202–208. [Google Scholar] [CrossRef]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Gribskov, M. Combining evidence using p-values: Application to sequence homology searches. Bioinformatics 1998, 14, 48–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; Van Der Lee, R.; Bessy, A.; Cheneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D260–D266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [Green Version]
- Fedotova, A.; Bonchuk, A.; Mogila, V.; Georgiev, P. C2H2 zinc finger proteins: The largest but poorly explored family of higher eukaryotic transcription factors. Acta Nat. 2017, 9, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Han, G.; Lu, C.; Guo, J.; Qiao, Z.; Sui, N.; Qiu, N.; Wang, B. C2H2 zinc finger proteins: Master regulators of abiotic stress responses in plants. Front. Plant Sci. 2020, 11, 115. [Google Scholar] [CrossRef] [Green Version]
- Shelest, E. Transcription factors in fungi: TFome dynamics, three major families, and dual-specificity TFs. FEMS Microbiol. Lett. 2008, 286, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Forde, B.G. AT-rich elements (ATREs) in the promoter regions of nodulin and other higher plant genes: A novel class of cis-acting regulatory element? Results Probl. Cell Differ. 1994, 20, 87–103. [Google Scholar] [CrossRef]
- Vardhanabhuti, S.; Wang, J.; Hannenhalli, S. Position and distance specificity are important determinants of cis-regulatory motifs in addition to evolutionary conservation. Nucleic Acids Res. 2007, 35, 3203–3213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iengar, P.; Joshi, N. Identification of putative regulatory motifs in the upstream regions of co-expressed functional groups of genes in Plasmodium falciparum. BMC Genom. 2009, 10, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Hou, L.; Su, N.; Hu, H.; Deng, M.; Li, X. Systematic identification of conserved motif modules in the human genome. BMC Genom. 2010, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magasanik, B.; Kaiser, C.A. Nitrogen regulation in Saccharomyces cerevisiae. Gene 2002, 290, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kageyama-Yahara, N.; Yamamichi, N.; Takahashi, Y.; Takeuchi, C.; Matsumoto, Y.; Sakaguchi, Y.; Koike, K. Tandem repeats of the 5′ flanking region of human MUC5AC have a role as a novel enhancer in MUC5AC gene expression. Biochem. Biophys. Rep. 2019, 18, 100632. [Google Scholar] [CrossRef] [PubMed]
- Vatov, E.; Ludewig, U.; Zentgraf, U. Disparate dynamics of gene body and cis-regulatory element evolution illustrated for the senescence-associated cysteine protease gene SAG12 of plants. Plants 2021, 10, 1380. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Gibson, S.; Williamson, D.; Rampling, D.; Austin, C.; Shipley, J.; Sebire, N.; Brock, P. Rapid and accurate determination of MYCN copy number and 1p deletion in neuroblastoma by quantitative PCR. Pediatr. Blood Cancer 2006, 46, 820. [Google Scholar] [CrossRef]
- Kim, T.-K.; Shiekhattar, R. Architectural and functional commonalities between enhancers and promoters. Cell 2015, 162, 948–959. [Google Scholar] [CrossRef] [Green Version]
- Tippens, N.D.; Vihervaara, A.; Lis, J.T. Enhancer transcription: What, where, when, and why? Genes Dev. 2018, 32, 1–3. [Google Scholar] [CrossRef]
- Serfling, E.; Lübbe, A.; Dorsch-Häsler, K.; Schaffner, W. Metal-dependent SV40 viruses containing inducible enhancers from the upstream region of metallothionein genes. EMBO J. 1985, 4, 3851–3859. [Google Scholar] [CrossRef]
- Goppelt, A.; Stelzer, G.; Lottspeich, F.; Meisterernst, M. A mechanism for repression of class II gene transcription through specific binding of NC2 to TBP-promoter complexes via heterodimeric histone fold domains. EMBO J. 1996, 15, 3105–3116. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.A.; Almada, A.E.; Zamudio, J.R.; Sharp, P.A. Antisense RNA polymerase II divergent transcripts are P-TEFb dependent and substrates for the RNA exosome. Proc. Natl. Acad. Sci. USA 2011, 108, 10460–10465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, F.; Fenouil, R.; Gut, M.; Cauchy, P.; Albert, T.K.; Zacarias-Cabeza, J.; Spicuglia, S.; De La Chapelle, A.L.; Heidemann, M.; Hintermair, C.; et al. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat. Struct. Mol. Biol. 2011, 18, 956–963. [Google Scholar] [CrossRef]
- Natoli, G.; Andrau, J.-C. Noncoding transcription at enhancers: General principles and functional models. Annu. Rev. Genet. 2012, 46, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Young, R.S.; Kumar, Y.; Bickmore, W.A.; Taylor, M.S. Bidirectional transcription initiation marks accessible chromatin and is not specific to enhancers. Genome Biol. 2017, 18, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scruggs, B.S.; Gilchrist, D.A.; Nechaev, S.; Muse, G.W.; Burkholder, A.; Fargo, D.C.; Adelman, K. Bidirectional transcription arises from two distinct hubs of transcription factor binding and active chromatin. Mol. Cell 2015, 58, 1101–1112. [Google Scholar] [CrossRef] [Green Version]
- Cooper, G.M.; Brown, C.D. Qualifying the relationship between sequence conservation and molecular function. Genome Res. 2008, 18, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Schoch, C.L.; Sung, G.H.; López-Giráldez, F.; Townsend, J.P.; Miadlikowska, J.; Hofstetter, V.; Robbertse, B.; Matheny, P.B.; Kauff, F.; Wang, Z.; et al. The Ascomycota tree of life: A phylum-wide phylogeny clarifies the origin and evolution of fundamental reproductive and ecological traits. Syst. Biol. 2009, 58, 224–239. [Google Scholar] [CrossRef]
- Spokevicius, A.; Taylor, L.; Melder, E.; Van Beveren, K.; Tibbits, J.; Creux, N.; Bossinger, G. The use of Induced Somatic Sector Analysis (ISSA) for studying genes and promoters involved in wood formation and secondary stem development. J. Vis. Exp. 2016, 116, e54553. [Google Scholar] [CrossRef]
- Duester, G. Knocking out enhancers to enhance epigenetic research. Trends Genet. 2019, 35, 89. [Google Scholar] [CrossRef]
- Borys, S.M.; Younger, S.T. Identification of functional regulatory elements in the human genome using pooled CRISPR screens. BMC Genom. 2020, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Sayari, M.; Van der Nest, M.A.; Steenkamp, E.T.; Adegeye, O.; Marincowitz, S.; Wingfield, B.D. Agrobacterium-mediated transformation of Ceratocystis albifundus. Microbiol. Res. 2019, 226, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.M.; Wingfield, B.D. CRISPR-cas9-mediated genome editing in the filamentous ascomycete Huntiella omanensis. J. Vis. Exp. 2020, 160, e61367. [Google Scholar] [CrossRef]
- Lane, F.A.; Du Plessis, D.; Wingfield, B.D.; Wilken, P.M. Transferring an Agrobacterium-mediated transformation protocol across eight genera in the Ceratocystidaceae. For. Pathol. 2021, 51, e12688. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Strain 1 | Whole Genome Accession Number | References 2 |
|---|---|---|---|
| Ceratocystidaceae | |||
| Ambrosiella beaveri | CMW 26179 * | JARQWA010000000 | Unpublished |
| Ambrosiella cleistominuta | CBS 141682 * | JABFIG010000000 | [20] |
| Ambrosiella xylebori | CBS 110.61 | PCDO01000000 | [21] |
| Berkeleyomyces basicola | CMW 25440 * | PJAC01000000 | [22] |
| Ceratocystis adiposa | CMW 2574 * | LXGU01000000 | [23] |
| Ceratocystis albifundus | CMW 17620 | JSSU01000000 | [24] |
| Ceratocystis fimbriata | CBS 114723 | APWK03000000 | [25] |
| Ceratocystis eucalypticola | CMW 9998 | LJOA01000000 | [26] |
| Ceratocystis harringtonii | CMW 14789 | MKGM01000000 | [27] |
| Ceratocystis manginecans | CMW 17570 | JJRZ01000000 | [28] |
| Ceratocystis smalleyi | CMW 14800 | NETT01000000 | [29] |
| Davidsoniella australis | CMW 2333 * | RHLR01000000 | [30] |
| Davidsoniella eucalypti | CMW 3254 * | RMBW01000000 | [31] |
| Davidsoniella neocaledoniae | CMW 26392 | RHDR01000000 | [30] |
| Davidsoniella virescens | CMW 17339 | LJZU01000000 | [26] |
| Endoconidiophora laricicola | CBS 100207 | LXGT01000000 | [23] |
| Endoconidiophora polonica | CBS 100205 | LXKZ01000000 | [23] |
| Huntiella bhutanensis | CMW 8217 | MJMS01000000 | [27] |
| Huntiella decipiens | CMW 30855 | NETU01000000 | [32] |
| Huntiella moniliformis | CBS 118127 | JMSH01000000 | [28] |
| Huntiella omanensis | CMW 11056 | JSUI01000000 | [24] |
| Huntiella savannae | CMW 17300 | LCZG01000000 | [33] |
| Thielaviopsis ethacetica | JCM 6961 | BCFY01000000 | [34] |
| Thielaviopsis musarum | CMW 1546 | LKBB01000000 | [26] |
| Thielaviopsis euricoi | JCM 6020 | BCHJ01000000 | [34] |
| non-Ceratocystidaceae | |||
| Neurospora crassa | OR74A | AABX03000000 | [35] |
| Sordaria macrospora | k-hell | CABT02000000 | [36] |
| Fusarium circinatum | FSP 34 | AYJV02000000 | [37] |
| Ophiostoma novo-ulmi | H327 | AMZD01000000 | [38] |
| BUSCO ID | Gene Name |
|---|---|
| EOG092D0072 | Dynein heavy chain (Penicillium sp. 2HH) |
| EOG092D00LL | Pre-mRNA-processing-splicing factor 8 (Penicillium sp. 2HH) |
| EOG092D0124 | General negative regulator of transcription subunit 1 (Penicillium sp. 2HH) |
| EOG092D01IY | SNF2-related protein (Penicillium sp. 2HH) |
| EOG092D01J4 | U3 snoRNP protein (Arthrobotrys oligospora) |
| EOG092D01MX | Phospholipase D family protein (Penicillium sp. 2HH) |
| EOG092D01QP | Cell morphogenesis protein PAG1 (Penicillium sp. 2HH) |
| EOG092D01WX | phosphatidylinositol-4- kinase (Arthrobotrys oligospora) |
| EOG092D01YA | Clathrin, heavy chain (Penicillium sp. 2HH) |
| EOG092D01ZK | E3 ubiquitin-protein ligase listerin (Penicillium sp. 2HH) |
| EOG092D02YC | Transcriptional regulatory protein sin3 (Arthrobotrys oligospora) |
| EOG092D03RC | UDP-glucose:glycoprotein glucosyltransferase (Penicillium sp. 2HH) |
| EOG092D03RY | Sister chromatid cohesion protein 2 (Arthrobotrys oligospora) |
| EOG092D042R | DNA repair protein rad50 (Arthrobotrys oligospora) |
| EOG092D0454 | Anaphase-promoting complex subunit 1 (Arthrobotrys oligospora) |
| EOG092D0564 | THO complex subunit 2 (Arthrobotrys oligospora) |
| EOG092D05RI | PAN2-PAN3 deadenylation complex catalytic subunit PAN2 (Penicillium sp. 2HH) |
| EOG092D05X9 | Transcription elongation factor spt6 (Helotiales sp. DMI_Dod_QoI) |
| EOG092D0ACX | Elongation factor EF-Tu (Penicillium sp. 2HH) |
| EOG092D0AI2 | MIFG and Upf2 domain-containing protein (Penicillium sp. 2HH) |
| Group Number | Genes (BUSCO ID) | Matched Motifs 1 |
|---|---|---|
| 1 | EOG092D01MX | Motif 28 |
| EOG092D00LL | Motif 1 | |
| EOG092D01YA | Motif 8 2 | |
| 2 | EOG092D00LL | Motif 2, Motif 3, Motif 5, Motif 6 |
| EOG092D03RY | Motif 18 | |
| 3 | EOG092D0124 | Motif 10 |
| EOG092D00LL | Motif 4 | |
| 4 | EOG092D0454 | Motif 22 2 |
| EOG092D0564 | Motif 2 2, Motif 3 2 | |
| EOG092D01YA | Motif 8 2, Motif 18 2 | |
| EOG092D01IY | Motif 9 2, Motif 4 2 | |
| EOG092D01WX | Motif 7 2 | |
| EOG092D0072 | Motif 1 2, Motif 4 2 | |
| EOG092D0124 | Motif 12 | |
| EOG092D03RC | Motif 9, Motif 14 |
| Known TF Classes | Number of Matched Motifs |
|---|---|
| C2H2 zinc finger factors | 151 |
| MADS box factors | 22 |
| Fork head/winged helix factors | 11 |
| Tryptophan cluster factors | 8 |
| Other | 7 |
| Helix–turn–helix | 6 |
| AP2/ERF domain | 5 |
| B3 domain | 3 |
| A.T hook factors | 2 |
| Heat shock factors | 2 |
| Basic helix–loop–helix factors (bHLH) | 2 |
| APSES-type DNA-binding domain | 1 |
| C6 zinc cluster factors | 1 |
| Other C4 zinc finger-type factors | 1 |
| Other α | 1 |
| Homeo domain factors | 1 |
| High-mobility group (HMG) domain factors | 1 |
| Basic leucine zipper (bZIP) | 1 |
| Copper-fist DNA-binding domain | 1 |
| Rel homology region (RHR) factors | 1 |
| Known TF Classes | Number of Matched Motifs |
|---|---|
| C2H2 zinc finger factors | 24 |
| Heat shock factors | 6 |
| Fork head/winged helix factors | 6 |
| Tryptophan cluster factor | 5 |
| Other C4 zinc finger-type factors | 5 |
| Basic helix–loop–helix factors (bHLH) | 4 |
| A.T Hook factors | 4 |
| C6 zinc cluster factors | 4 |
| Copper-first DNA-binding domain | 3 |
| High-mobility group (HMG) domain factors | 3 |
| Heteromeric CCAAT-binding | 2 |
| Basic leucine zipper factors (bZIP) | 2 |
| Homeo domain factors | 2 |
| MADS box factors | 1 |
| APSES-type DNA-binding domain | 1 |
| Species | Motif |
|---|---|
| EOG092D0072 | |
| N. crassa | Motif 1 |
| F. circinatum | Motif 4 |
| O. novo-ulmi | Motif 1, Motif 4 |
| S. macrospora | Motif 1 |
| EOG092D01ZK | |
| F. circinatum | Motif 3 |
| O. novo-ulmi | Motif 2, Motif 3 |
| EOG092D01J4 | |
| F. circinatum | Motif 6 |
| EOG092D01YA | |
| N. crassa | Motif 1, Motif 2 |
| F. circinatum | Motif 2, Motif 5, Motif 9, Motif 18 |
| O. novo-ulmi | Motif 2, Motif 5, Motif 8, Motif 18 |
| S. macrospora | Motif 3, Motif 5, Motif 8 |
| EOG092D01WX | |
| F. circinatum | Motif 1, Motif 2, Motif 4 |
| O. novo-ulmi | Motif 1, Motif 2, Motif 4, Motif 7 |
| S. macrospora | Motif 4, Motif 9 |
| EOG092D01MX | |
| N. crassa | Motif 28 |
| EOG092D01IY | |
| N. crassa | Motif 4 |
| O. novo-ulmi | Motif 4, Motif 9 |
| S. macrospora | Motif 4 |
| EOG092D0124 | |
| N. crassa | Motif 1 |
| O. novo-ulmi | Motif 1 |
| S. macrospora | Motif 1 |
| EOG092D0454 | |
| F. circinatum | Motif 22 |
| O. novo-ulmi | Motif 22 |
| EOG092D03RY | |
| O. novo-ulmi | Motif 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maseko, N.N.; Steenkamp, E.T.; Wingfield, B.D.; Wilken, P.M. An in Silico Approach to Identifying TF Binding Sites: Analysis of the Regulatory Regions of BUSCO Genes from Fungal Species in the Ceratocystidaceae Family. Genes 2023, 14, 848. https://doi.org/10.3390/genes14040848
Maseko NN, Steenkamp ET, Wingfield BD, Wilken PM. An in Silico Approach to Identifying TF Binding Sites: Analysis of the Regulatory Regions of BUSCO Genes from Fungal Species in the Ceratocystidaceae Family. Genes. 2023; 14(4):848. https://doi.org/10.3390/genes14040848
Chicago/Turabian StyleMaseko, Nomaswazi N., Emma T. Steenkamp, Brenda D. Wingfield, and P. Markus Wilken. 2023. "An in Silico Approach to Identifying TF Binding Sites: Analysis of the Regulatory Regions of BUSCO Genes from Fungal Species in the Ceratocystidaceae Family" Genes 14, no. 4: 848. https://doi.org/10.3390/genes14040848