
Phenotypes and Genotypes in Patients with SMC1A-Related Developmental and Epileptic Encephalopathy

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
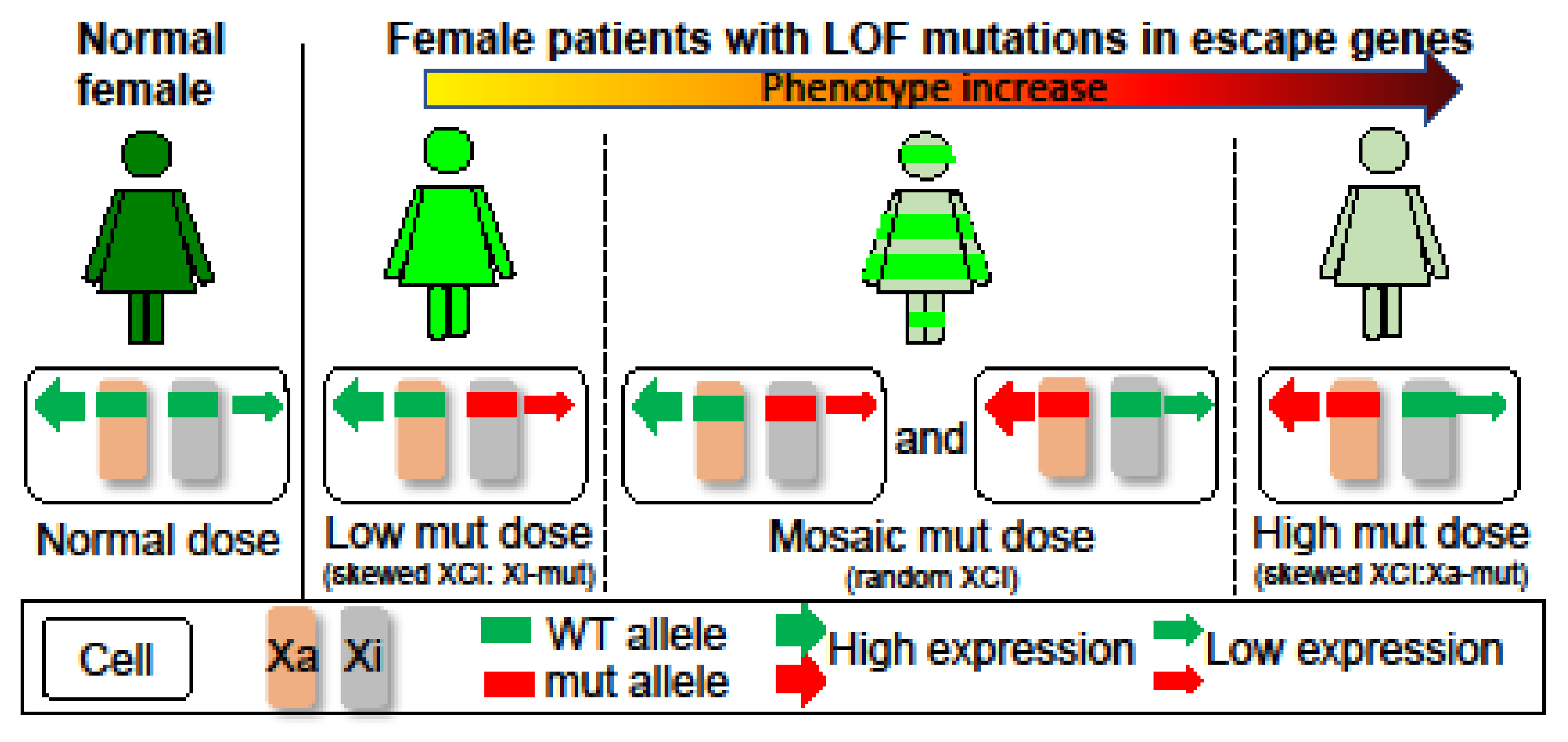
4. Discussion
5. Conclusions
6. Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Miller, A.P.; Carrel, L.; Rupert, J.L.; Davies, K.E.; Willard, H.F. The DXS423E gene in Xp11.21 escapes X chromosome inactivation. Hum. Mol. Genet. 1995, 4, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Carrel, L.; Willard, H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Balaton, B.P.; Cotton, A.M.; Brown, C.J. Derivation of consensus inactivation status for X-linked genes from genome-wide studies. Biol. Sex Differ. 2015, 6, 35. [Google Scholar] [CrossRef] [Green Version]
- Parenti, I.; Rovina, D.; Masciadri, M.; Cereda, A.; Azzollini, J.; Picinelli, C.; Limongelli, G.; Finelli, P.; Selicorni, A.; Russo, S.; et al. Overall and allele-specific expression of the SMC1A gene in female Cornelia de Lange syndrome patients and healthy controls. Epigenetics 2014, 9, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Davidson, I.F.; Peters, J.-M. Genome folding through loop extrusion by SMC complexes. Nat. Rev. Mol. Cell Biol. 2021, 22, 445–464. [Google Scholar] [CrossRef]
- Oldenkamp, R.; Rowland, B.D. A walk through the SMC cycle: From catching DNAs to shaping the genome. Mol. Cell 2022, 82, 1616–1630. [Google Scholar] [CrossRef] [PubMed]
- Selicorni, A.; Mariani, M.; Lettieri, A.; Massa, V. Cornelia de Lange Syndrome: From a Disease to a Broader Spectrum. Genes 2021, 12, 1075. [Google Scholar] [CrossRef] [PubMed]
- Huisman, S.; Mulder, P.A.; Redeker, E.; Bader, I.; Bisgaard, A.-M.; Brooks, A.; Cereda, A.; Cinca, C.; Clark, D.; Cormier-Daire, V.; et al. Phenotypes genotypes in individuals with SMC1A variants. Am. J. Med. Genet. A 2017, 173, 2108–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, M.I.; Jespersgaard, C.; Brøndum-Nielsen, K.; Bisgaard, A.-M.; Tümer, Z. Cornelia de Lange syndrome. Clin. Genet. 2015, 88, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Feldman, R.; Zhang, Z.; Deardorff, M.A.; Haverfield, E.V.; Kaur, M.; Li, J.R.; Clark, D.; Kline, A.D.; Waggoner, D.J.; et al. SMC1A expression and mechanism of pathogenicity in probands with X-Linked Cornelia de Lange syndrome. Hum. Mutat. 2009, 30, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.H.R.; Tim-Aroon, T.; Shieh, J.; Merrill, M.; Deeb, K.K.; Zhang, S.; Bass, N.E.; Bedoyan, J.K. Novel SMC1A frameshift mutations in children with developmental delay and epilepsy. Eur. J. Med. Genet. 2015, 58, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.; Kleefstra, T.; Willemsen, M.H.; de Vries, P.; Pfundt, R.; Hehir-Kwa, J.Y.; Gilissen, C.; Veltman, J.A.; de Vries, B.B.A.; Vissers, L.E.L.M. De novo loss-of-function mutations in X-linked SMC1A cause severe ID and therapy-resistant epilepsy in females: Expanding the phenotypic spectrum. Clin. Genet. 2016, 90, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, N.; Lebon, S.; Jeannet, P.-Y.; Jacquemont, S.; Billuart, P.; Bienvenu, T. Early-onset encephalopathy with epilepsy associated with a novel splice site mutation in SMC1A. Am. J. Med. Genet. A 2015, 167A, 3076–3081. [Google Scholar] [CrossRef] [PubMed]
- Symonds, J.D.; Joss, S.; Metcalfe, K.A.; Somarathi, S.; Cruden, J.; Devlin, A.M.; Donaldson, A.; DiDonato, N.; Fitzpatrick, D.; Kaiser, F.J.; et al. Heterozygous truncation mutations of the SMC1A gene cause a severe early onset epilepsy with cluster seizures in females: Detailed phenotyping of 10 new cases. Epilepsia 2017, 58, 565–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorman, K.M.; Forman, E.; Conroy, J.; Allen, N.M.; Shahwan, A.; Lynch, S.A.; Ennis, S.; King, M.D. Novel SMC1A variant and epilepsy of infancy with migrating focal seizures: Expansion of the phenotype. Epilepsia 2017, 58, 1301–1302. [Google Scholar] [CrossRef] [Green Version]
- Oguni, H.; Nishikawa, A.; Sato, Y.; Otani, Y.; Ito, S.; Nagata, S.; Kato, M.; Hamanaka, K.; Miyatake, S.; Matsumoto, N. A missense variant of SMC1A causes periodic pharmaco-resistant cluster seizures similar to PCDH19-related epilepsy. Epilepsy Res. 2019, 155, 106149. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Zhang, X.; Xiao, B.; Zhang, L.; Long, H. A de novo mutation in SMC1A gene identified in a Chinese infant with nonclassical Cornelia de Lange syndrome and drug-resistant epilepsy. Neurol. Sci. 2021, 42, 329–331. [Google Scholar] [CrossRef]
- Barañano, K.W.; Kimball, A.; Fong, S.L.; Egense, A.S.; Hudon, C.; Kline, A.D. Further Characterization of SMC1A Loss of Function Epilepsy Distinct From Cornelia de Lange Syndrome. J. Child. Neurol. 2022, 37, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Elwan, M.; Fowkes, R.; Lewis-Smith, D.; Winder, A.; Baker, M.R.; Thomas, R.H. Late-onset cluster seizures and intellectual disability associated with a novel truncation variant in SMC1A. Epilepsy Behav. Rep. 2022, 19, 100556. [Google Scholar] [CrossRef]
- Wenger, T.L.; Chow, P.; Randle, S.C.; Rosen, A.; Birgfeld, C.; Wrede, J.; Javid, P.; King, D.; Manh, V.; Hing, A.V.; et al. Novel findings of left ventricular non-compaction cardiomyopathy, microform cleft lip and poor vision in patient with SMC1A-associated Cornelia de Lange syndrome. Am. J. Med. Genet. A 2017, 173, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Mulder, P.A.; Huisman, S.; Landlust, A.M.; Moss, J.; SMC1A Consortium; Piening, S.; Hennekam, R.C.; van Balkom, I.D.C. Development, behaviour and autism in individuals with SMC1A variants. J. Child. Psychol. Psychiatry 2019, 60, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Kruszka, P.; Berger, S.I.; Casa, V.; Dekker, M.R.; Gaesser, J.; Weiss, K.; Martinez, A.F.; Murdock, D.R.; Louie, R.J.; Prijoles, E.J.; et al. Cohesin complex-associated holoprosencephaly. Brain. J. Neurol. 2019, 142, 2631–2643. [Google Scholar] [CrossRef] [Green Version]
- Chinen, Y.; Nakamura, S.; Kaneshi, T.; Nakayashiro, M.; Yanagi, K.; Kaname, T.; Naritomi, K.; Nakanishi, K. A novel nonsense SMC1A mutation in a patient with intractable epilepsy and cardiac malformation. Hum. Genome Var. 2019, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Spagnoli, C.; Fusco, C.; Pisani, F. Rett Syndrome Spectrum in Monogenic Developmental-Epileptic Encephalopathies and Epilepsies: A Review. Genes 2021, 12, 1157. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.C.; Zoghbi, H.Y.; Moseley, A.B.; Rosenblatt, H.M.; Belmont, J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992, 51, 1229–1239. [Google Scholar]
- Machado, F.B.; Machado, F.B.; Faria, M.A.; Lovatel, V.L.; Alves da Silva, A.F.; Radic, C.P.; De Brasi, C.D.; Rios, Á.F.L.; de Sousa Lopes, S.M.C.; da Silveira, L.S.; et al. 5meCpG epigenetic marks neighboring a primate-conserved core promoter short tandem repeat indicate X-chromosome inactivation. PLoS ONE 2014, 9, e103714. [Google Scholar] [CrossRef] [PubMed]
- Deardorff, M.A.; Kaur, M.; Yaeger, D.; Rampuria, A.; Korolev, S.; Pie, J.; Gil-Rodríguez, C.; Arnedo, M.; Loeys, B.; Kline, A.D.; et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet. 2007, 80, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, M.; Scheinost, J.C.; Petela, N.J.; Gligoris, T.G.; Wissler, M.; Ogushi, S.; Collier, J.E.; Voulgaris, M.; Kurze, A.; Chan, K.-L.; et al. The Cohesin Ring Uses Its Hinge to Organize DNA Using Non-topological as well as Topological Mechanisms. Cell 2018, 173, 1508–1519.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tierney, A.L.; Nelson, C.A. Brain Development and the Role of Experience in the Early Years. Zero Three 2009, 30, 9–13. [Google Scholar] [PubMed]
- Fujita, Y.; Masuda, K.; Bando, M.; Nakato, R.; Katou, Y.; Tanaka, T.; Nakayama, M.; Takao, K.; Miyakawa, T.; Tanaka, T.; et al. Decreased cohesin in the brain leads to defective synapse development and anxiety-related behavior. J. Exp. Med. 2017, 214, 1431–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.; Deng, X.; Disteche, C.M. X-factors in human disease: Impact of gene content and dosage regulation. Hum. Mol. Genet. 2021, 30, R285–R295. [Google Scholar] [CrossRef] [PubMed]
- Moncayo, J.A.; Ayala, I.N.; Argudo, J.M.; Aguirre, A.S.; Parwani, J.; Pachano, A.; Ojeda, D.; Cordova, S.; Mora, M.G.; Tapia, C.M.; et al. Understanding Protein Protocadherin-19 (PCDH19) Syndrome: A Literature Review of the Pathophysiology. Cureus 2022, 14, e25808. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Cobos, M.J.; Balaton, B.P.; Brown, C.J. Genes that escape from X-chromosome inactivation: Potential contributors to Klinefelter syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Berletch, J.B.; Ma, W.; Yang, F.; Shendure, J.; Noble, W.S.; Disteche, C.M.; Deng, X. Escape from X inactivation varies in mouse tissues. PLoS Genet. 2015, 11, e1005079. [Google Scholar] [CrossRef]
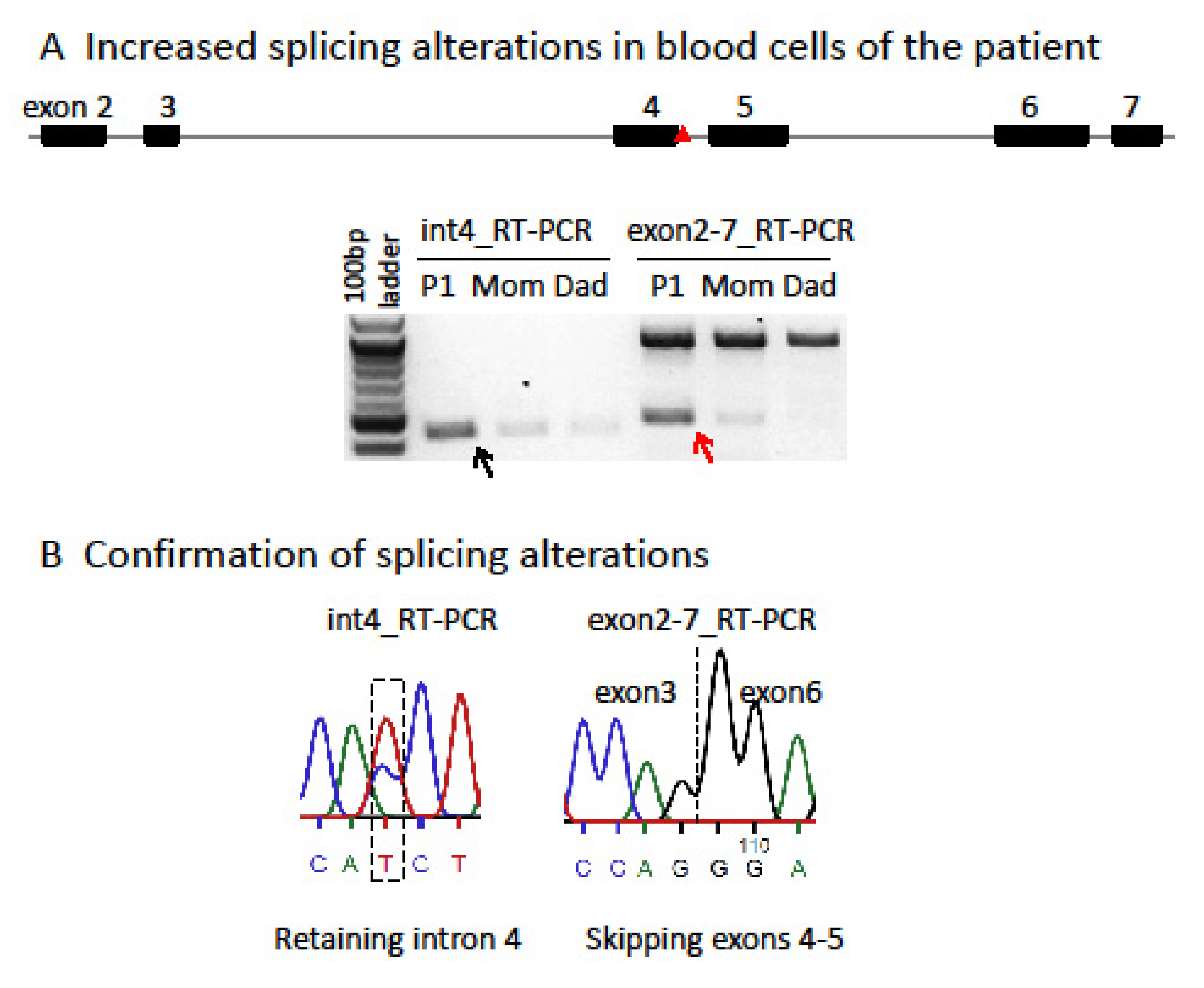

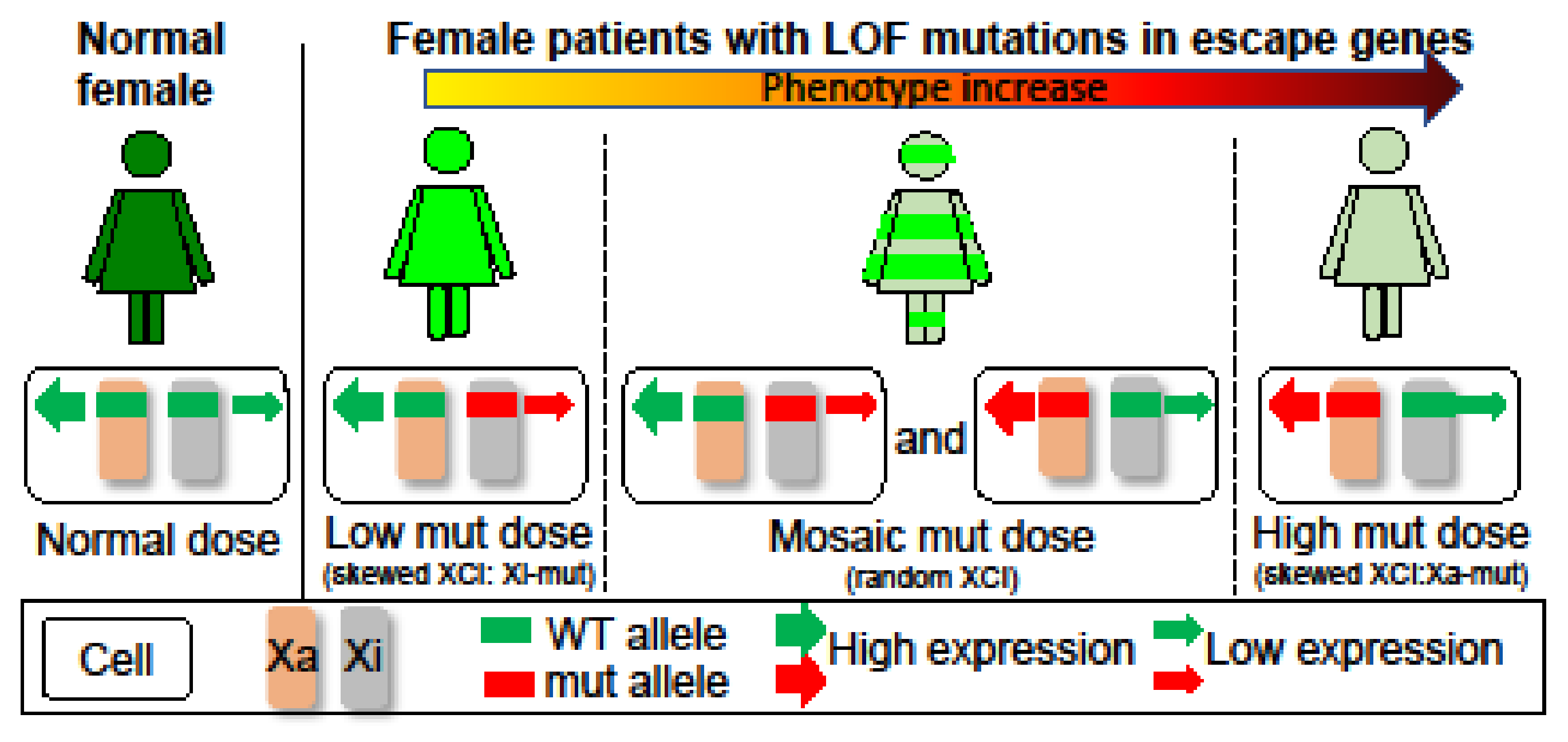

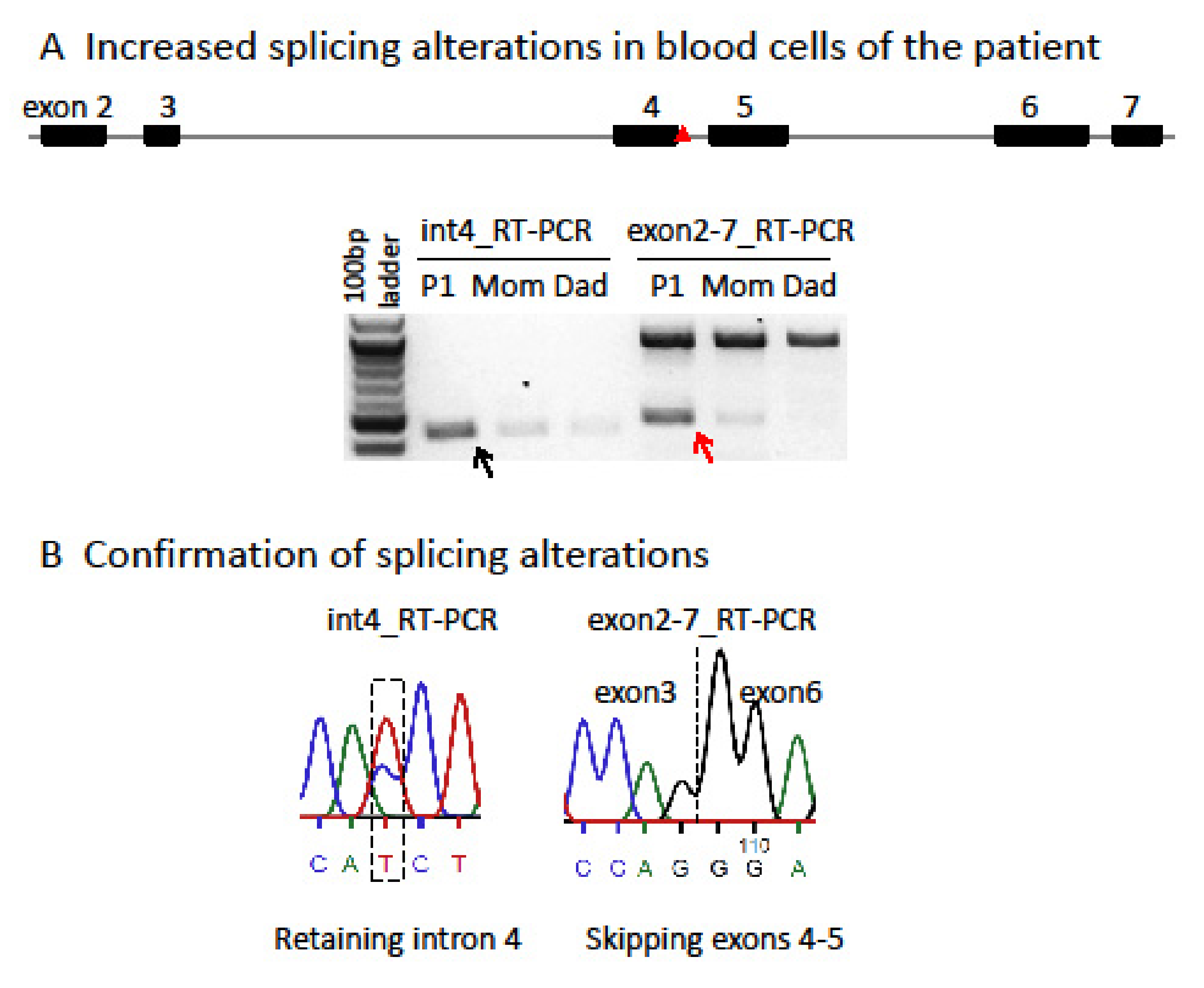{kind=link}
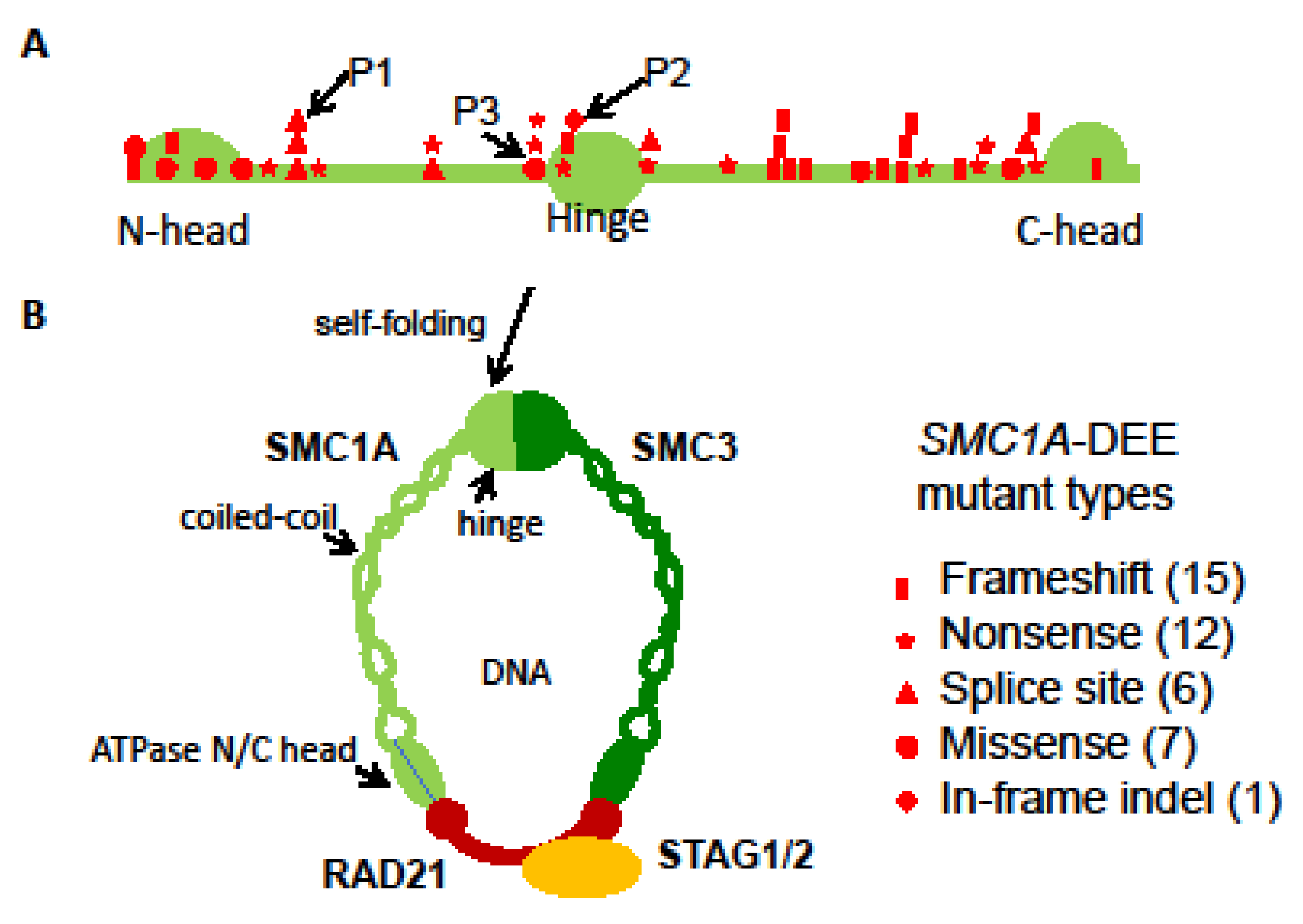{kind=link}
{kind=link}
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Full term | Yes | Yes | No, 36 + 6/7 wk |
| Congenital microcephaly | No | No | Yes |
| Postnatal microcephaly | Yes | Yes | Yes initially |
| Hypotonia | Yes | Yes | Yes |
| Expressive language | None | None | A few words |
| Encephalopathy | Profound | Profound | Severe |
| G-tube | Yes (4 yr) | Yes (9 M) | No |
| CDLS features | No | Minor | Minor |
| Independent walking | No | No | Yes |
| Precocious puberty | Yes | No | No |
| Cardiac abnormalities | No | Left ventricular cardiomyopathy | No |
| MRI: | A small cyst in left anterior temporal region (age 7 M and 9 yr) | Normal (2 yr) | No |
| Patient 1 | Patient 2 | Patient 3 | |
|---|---|---|---|
| Age of seizure onset (M) | 4 | 2 | 35 |
| Intractable epilepsy | Yes | Yes | Yes |
| Epilepsy syndrome | |||
| LGS | Yes | No | Yes |
| Epileptic spasms | No | Yes (9 M) | No |
| History of status epilepticus | Yes | Yes | No |
| Seizure burden | Weekly and up to daily | Weekly and up to daily | Less than monthly |
| Seizure Types | |||
| Tonic/atonic | Yes | Yes | Yes |
| Tonic clonic | Yes | Yes | Yes |
| Myoclonic | Yes | No | Yes |
| Focal onset, motor | Yes | Yes | No |
| Focal onset, nonmotor | Yes | Yes | No |
| Atypcial absence | Yes | No | No |
| Cluster seizures | Yes | Yes | Yes |
| Age starting AED | 4 M | 2M | 47M |
| Current AED: | Topiramate, Perampanel, Epidiolex, Lacosamide | Clobazam Levetiracetam | Brivaracetam Rufinamide Clobazam |
| Past AED | Zonisamide, Valproic acid, Clobazam Oxcarbazepine, Levetiracetam, Rufinamide, Lamotrigene, Brivaracetam | Phenobarbital, Oxcarbazepine, ACTH, Viagabatrin, Diazepam, Zonisamide | Valproci acid, Levetiracetam |
| AEDs: severe adverse effects | No | No | No |
| Ketogenic/modified Atkin’s diet | Yes, no efficacy | Yes, partial efficacy | Yes, no efficacy |
| Vagal nerve stimulator | Yes, unclear efficacy | No | No |
| Epilepsy resection surgery | No | No | No |
| EEG | |||
| Normal Yes/No | No | No | No |
| Posterior dominant rhythm | Yes, slow for age | No | Yes, slow for age |
| EEG background | Diffuse slow | Diffuse slow, history of hypsarrhythmia | Diffuse slow |
| Sleep architecture | No | No | Yes |
| Interictal pattern | Generalized; multifocal, more posteriorly | Multifocal | Generalized spike wave |
| GPFA | Yes | No | No |
| Slow Spike wave | No | No | Yes |
| Variant # | Nucleotide Changes | Amino Acid Changes | Domain | Type | XCI | Ref. | Age of Sz Onset | Speech | ID | Walking | MRI |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | C.20_23del | Ile7Argfs*42 | P1 head | Frameshift | Moderately skewed 81:19 | Barañano, 2022 [19] (P6) | 15 M | none | yes | No | n/a |
| 2 | c.31A>T | Asn11Tyr | P1 head | Missense | n/a | Huisman, 2017 [9] (P1) | 2.5 M | n/a | yes | normal | |
| 3 | c.140T>G | Phe47Cys | P1 head | Missense | Random 74:26 | Barañano, 2022 [19] (P3) | 3 M | none | yes | No | n/a |
| 4 | c.157dup | Thr53AsnfsX34 | P1 head | Frameshift | n/a | Huisman, 2017 [9] (P2) | 5 M | n/a | yes | n/a | cerebral volume loss |
| 5 | c.287G>C | Arg96Pro | P1 head | Missense | Random | Barañano, 2022 [19] (P11) | 18 M | n/a | yes | n/a | n/a |
| 6 | c.421G>A | Glu141Lys | P1 head | Missense | highly Skewed 100:0 | Barañano, 2022 [19] (P1) | 2.5 M | none | yes | n/a | n/a |
| 7 | c.511C>T | Arg171Ter | Coil | Nonsense | Random | Symonds, 2017 [15] (P7) | 4 Weeks | none | yes | No | normal |
| 8 | c.615G>A | Glu205fs* | Coil | Splice-site | N/A | (NM:00128146:c.549G>A) in Symonds, 2017 [15] (P3); P12 in Barañano, 2022 [19] | P3 Symonds-4 M; P12 Barañano-13 M | none | yes | No | P3 Symonds-small hemorrhage along posterior falx and tentorium; P12-Barañano-n/a |
| 9 | c.615G+1G>C | Glu205fs* | Coil | Splice-site | Moderately skewed 83:17 | Barañano, 2022 [19] (P9) | 1 M | n/a | yes | No | n/a |
| 10 (P1 in our cohort) | c.615+5G>A | Glu205fs* | Coil | Splice-site | Random | Our cohort P1 | 4 M | none | yes | no | normal |
| 11 | c.694G>T | Glu232Ter | Coil | Nonsense | n/a | Huisman, 2017 [9] (P7) | 4 M | n/a | yes | n/a | n/a |
| 12 | c.1113+1G>A | Gln371fs* | Coil | Splice-site | n/a | Gorman 2017 [16] (P2) | 7 Weeks | none | yes | n/a | n/a |
| 13 | c.1114delG | Val372Ter | Coil | Nonsense | n/a | Gorman 2017 [16] (P1) | 7 Weeks | none | yes | no | n/a |
| 14 (P3 in our cohot) | c.1487G>A | Arg496His | Close to hinge | Missense | Skewed in our patient P3; N/A in 5 patients from Deardorff | Our cohort P3; Deardorff, 2007 [28] (P3) | 35 M | limited | yes | Yes | n/a |
| 15 | c.1489C>T | Arg497Ter | Close to hinge | Nonsense | n/a | Fang, 2021 [18] | 4 M | none | yes | No | normal |
| 16 | c.1495C>T | Arg499Ter | Close to hinge | Nonsense | n/a | Chinen, 2019 [24]; Kruszka, 2019 [23] (P8) | 40 M-Chinen, 2019; N/A- | none | yes | Chinen-Yes | ChinenP8- microform of HPE, Kruszka P8-Triventricular ectasia |
| 17 | c.1591C>T | Gln531Ter | Hinge | Nonsense | Moderately skewed | Symonds, 2017 [15] (P1) | 15 M | none | yes | Yes | normal |
| 18 | c.1609delG | Val537Phefs*42 | Hinge | Frameshift | highly skewed | Barañano, 2022 [19] (P8) | 5 M | none | yes | No | n/a |
| 19 (P2 in our cohort) | c.1636_1638delATT | Ilu546del | Hinge | In-frame | Random | reported in Wenger, 2016 [21] | 2 M | none | yes | No | normal |
| 20 | c.1900C>T | Gln634Ter | Close to hinge | Nonsense | Moderately skewed 86:14, Mosaic | Barañano, 2022 [19] (P2) | 3 M | n/a | yes | n/a | n/a |
| 21 | c.1911+1G>T | Thr638Valfs*48 | Close to hinge | Splice-site | n/a | Lebrun, 2015 [14] | Neonate | none | yes | no | small frontal lobe, thin CC |
| 22 | c.2197G>T | Glu733Ter | Coil | Nonsense | Random | Symonds, 2017 [15] (P4) | 5 M | none | yes | No | volume loss |
| 23 | c.2364del | Asn788Lysfs*10 | Coil | Frameshift | Random | Jansen, 2016 [16] (P1) | 9 M | none | yes | No | slightly enlarged ventricles, hypotrophy cerebellar vermis |
| 24 | c.2394delA | Lys798Asnfs*31 | Coil | Frameshift | n/a | Kruszka, 2019 [23] (P9) | n/a | n/a | yes | semi-lobar HPE | |
| 25 | c.2394dupA | Arg799fs | Coil | Frameshift | n/a | Barañano, 2022 [19] (P5) | 4 M | none | yes | No | n/a |
| 26 | c.2421_2562del | Leu808Arg-fs*6 | Coil | Frameshift | Moderately skewed 85:15 | Jansen, 2016 [16] (P2) | 2 M | none | yes | None | mild periventricular white matter abnormalities |
| 27 | c.2477delA | p825fs | Coil | Frameshift | n/a | Symonds, 2017 [15] (P8, P9) | 28 M in P8; <1 M in P9 | P8-none; P9-none | yes | P8-yes | P8-normal; P9-hemi-lobar HPE |
| 28 | c.2683C>G | Arg895Gly | Coil | Missense | Skewed-P1; n/a-P2, P9 from Kruszka | Oguni, 2019 [17]; Kruszka, 2019 [23] (P9) | 25 M-P1 Oguni, P2 (mother of P1)-Oguni- 12y; n/a- P8-Kruszka | Oguin-none; Kruszka-n/a | P1-yes; P2-normal until sz onset | Oguni-P1-no, P2-yes, Kruszka-n/a | P2: cerebellar atrophy; P9-Kruszka-semi-lobar HPE |
| 29 | c.2769dupC | Ser924Glnfs*2 | Coil | Frameshift | n/a | Barañano, 2022 [19] (P10) | 24 M | no | yes | n/a | n/a |
| 30 | c.2834delG | Gly945Alafs*19 | Coil | Frameshift | n/a | Kruszka, 2019 [23] (P11) | n/a | n/a | yes | n/a | semi-lobar HPE |
| 31 | c.2853_2856delTCAG | Ser951Argfs*12 | Coil | Frameshift | Skewed | Goldstein, 2015 [12] (P1) | 4 M | n/a | severe | no | mild ventriculomegaly, mildly rotated hippocampal heads |
| 32 | c.2873delA | Gln958Argfs*6 | Coil | Frameshift | Random | Barañano, 2022 [19] (P7) | 3 M | none | Severe | no | n/a |
| 33 | c.2923C>T | Arg975Ter | Coil | Nonsense | N/A | Symonds, 2017 [15] (P6) | 5 M | Lost speech after status | Moderate to severe | yes | normal |
| 34 | c.3046_3048delGTGinsG | Val1016Alafs*28 | Coil | Frameshift | Random | Barañano, 2022 [19] (P4) | Neonate | n/a | yes | no | n/a |
| 35 | c.3115C>T | Gln1039Ter | Coil | Nonsense | Random 76:24 | Symonds, 2017 [15] (P10) | 2 M | none | yes | no | thin abnormally shaped CC and minimal cerebral atrophy |
| 36 | c.3145C>T | Arg1049Ter | Coil | Nonsense | n/a | Symonds, 2017 [15] (P2) | 5–6 weeks | none | yes | no | cerebral volume loss |
| 37 | c.3241A>T | Ile1081Phe | Close to P2 head | Missense | n/a | Unreported | 4 yr | none | yes | yes | mildly prominent lateral ventricles |
| 38 | c.3285+1G>C | p1095 | Close to P2 head | Splice-site | n/a | Kruszka, 2019 [23] (P7) | not reported | n/a | yes | n/a | middle interhemispheric variant, HPE |
| 39 | c.3312C>A | Tyr1107Ter | Close to P2 head | Nonsense | n/a | Elwan, 2022 [20] | 12 yr | normal | normal before SE | yes | normal |
| 40 | c.3326_3330delATGGCinsC | Asp1109Alafs*102 | Close to P2 head | Frameshift | n/a | Symonds, 2017 [15] (P5) | 6 M | none | yes | no | small cavum septum vergae |
| 41 | c.3549_3552dupGGCC | Ile1185glyfs*23 | P2 head | Frameshift | Random in patient 1-Goldstein, 2015; n/a-P3- Barañano, 2022 | Goldstein, 2015 [12] (P2); Barañano, 2022 [19] (P13) | 17 M- P2- Goldstein 2015 [12]; 16 M- P13-Barañano 2022 [19] | n/a-Goldstein; P13-Barañano-limited | yes | n/a-Goldstein; P13Barababi-yes | P2 Goldstein-mild enlarged extra-axial spaces and slight thinning of CC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozarth, X.L.; Lopez, J.; Fang, H.; Lee-Eng, J.; Duan, Z.; Deng, X. Phenotypes and Genotypes in Patients with SMC1A-Related Developmental and Epileptic Encephalopathy. Genes 2023, 14, 852. https://doi.org/10.3390/genes14040852
Bozarth XL, Lopez J, Fang H, Lee-Eng J, Duan Z, Deng X. Phenotypes and Genotypes in Patients with SMC1A-Related Developmental and Epileptic Encephalopathy. Genes. 2023; 14(4):852. https://doi.org/10.3390/genes14040852
Chicago/Turabian StyleBozarth, Xiuhua L., Jonathan Lopez, He Fang, Jacqueline Lee-Eng, Zhijun Duan, and Xinxian Deng. 2023. "Phenotypes and Genotypes in Patients with SMC1A-Related Developmental and Epileptic Encephalopathy" Genes 14, no. 4: 852. https://doi.org/10.3390/genes14040852
APA StyleBozarth, X. L., Lopez, J., Fang, H., Lee-Eng, J., Duan, Z., & Deng, X. (2023). Phenotypes and Genotypes in Patients with SMC1A-Related Developmental and Epileptic Encephalopathy. Genes, 14(4), 852. https://doi.org/10.3390/genes14040852