
Leri–Weill Dyschondrosteosis Caused by a Leaky Homozygous SHOX Splice-Site Variant

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
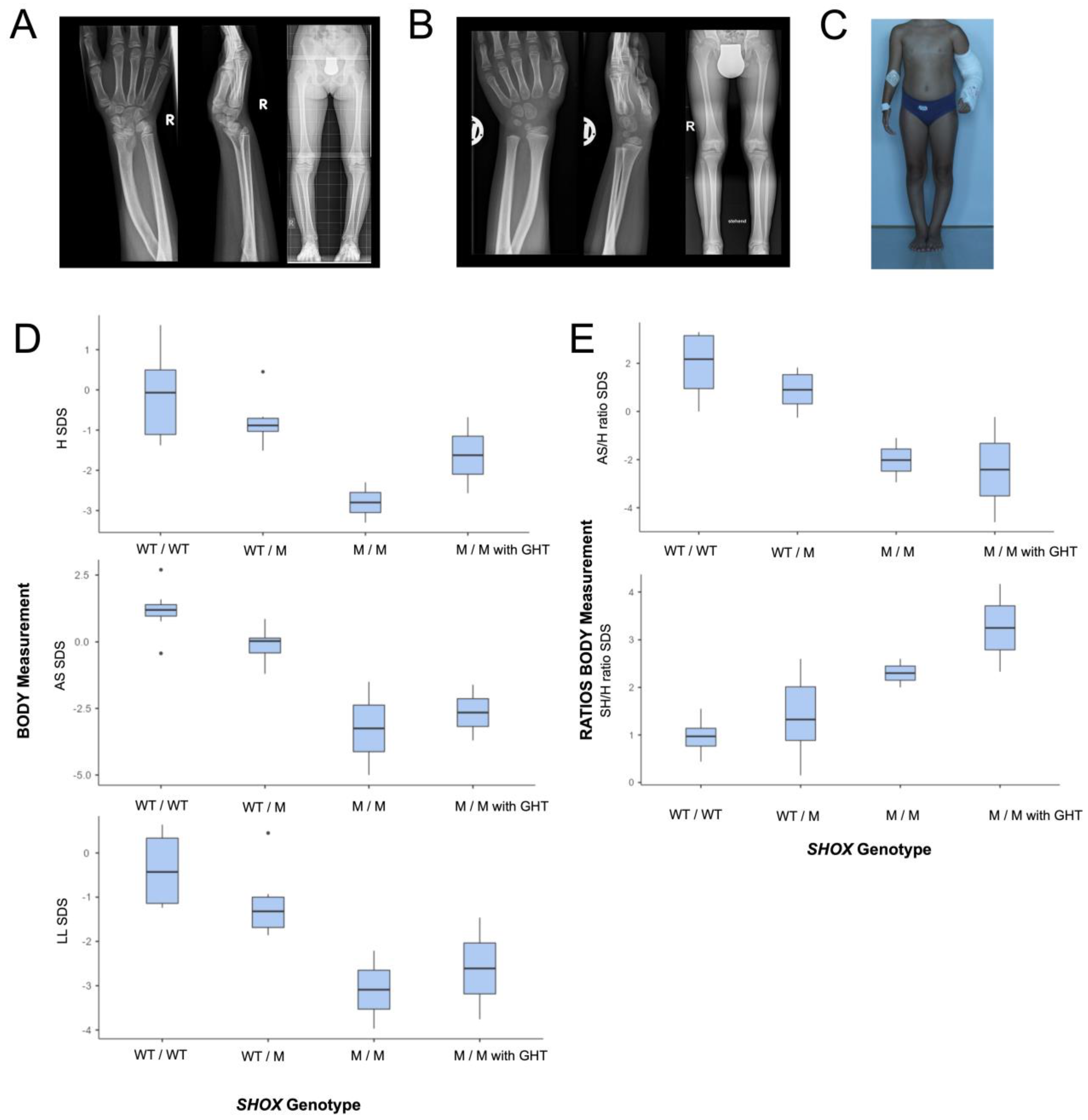
:1. Introduction
2. Materials and Methods
2.1. Ethics and Patient Samples
2.2. Clinical Assessment
2.3. Detection of SHOX Variants
2.4. SHOX Transcript Analysis
3. Results
3.1. Clinical Characteristics
3.1.1. Patients 1 and 2
3.1.2. Clinical Investigations and Body Measurements in the Whole Pedigree
3.2. Identification of a Novel Homozygous Splice-Site SHOX Variant
3.3. The SHOX Variant Represents a Leaky Splice Donor Site Mutation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rao, E.; Weiss, B.; Fukami, M.; Rump, A.; Niesler, B.; Mertz, A.; Muroya, K.; Binder, G.; Kirsch, S.; Winkelmann, M.; et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat. Genet. 1997, 16, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Marttila, T.; Winter, A.; Caldeira, S.; Malanchi, I.; Blaschke, R.J.; Häcker, B.; Rao, E.; Karperien, M.; Wit, J.M.; et al. The short stature homeodomain protein SHOX induces cellular growth arrest and apoptosis and is expressed in human growth plate chondrocytes. J. Biol. Chem. 2004, 279, 37103–37114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, G.; Rappold, G.A. SHOX Deficiency Disorders. In GeneReviews((R)); Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Shears, D.J.; Vassal, H.J.; Goodman, F.R.; Palmer, R.W.; Reardon, W.; Superti-Furga, A.; Scambler, P.J.; Winter, R.M. Mutation and deletion of the pseudoautosomal gene SHOX cause Leri-Weill dyschondrosteosis. Nat. Genet. 1998, 19, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, A.M.; Aksglaede, L.; Garn, I.; Tartaglia, N.; Tassone, F.; Gravholt, C.H.; Bojesen, A.; Sorensen, K.; Jorgensen, N.; Rajpert-De Meyts, E.; et al. Increased number of sex chromosomes affects height in a nonlinear fashion: A study of 305 patients with sex chromosome aneuploidy. Am. J. Med. Genet. A 2010, 152A, 1206–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, G. Short stature due to SHOX deficiency: Genotype, phenotype, and therapy. Horm. Res. Paediatr. 2011, 75, 81–89. [Google Scholar] [CrossRef]
- Belin, V.; Cusin, V.; Viot, G.; Girlich, D.; Toutain, A.; Moncla, A.; Vekemans, M.; Le Merrer, M.; Munnich, A.; Cormier-Daire, V. SHOX mutations in dyschondrosteosis (Leri-Weill syndrome). Nat. Genet. 1998, 19, 67–69. [Google Scholar] [CrossRef]
- Munns, C.F.; Glass, I.A.; Flanagan, S.; Hayes, M.; Williams, B.; Berry, M.; Vickers, D.; O’Rourke, P.; Rao, E.; Rappold, G.A.; et al. Familial growth and skeletal features associated with SHOX haploinsufficiency. J. Pediatr. Endocrinol. Metab. 2003, 16, 987–996. [Google Scholar] [CrossRef]
- Benabbad, I.; Rosilio, M.; Child, C.J.; Carel, J.C.; Ross, J.L.; Deal, C.L.; Drop, S.L.; Zimmermann, A.G.; Jia, N.; Quigley, C.A.; et al. Safety Outcomes and Near-Adult Height Gain of Growth Hormone-Treated Children with SHOX Deficiency: Data from an Observational Study and a Clinical Trial. Horm. Res. Paediatr. 2017, 87, 42–50. [Google Scholar] [CrossRef]
- Blum, W.F.; Crowe, B.J.; Quigley, C.A.; Jung, H.; Cao, D.; Ross, J.L.; Braun, L.; Rappold, G.; Group, S.S. Growth hormone is effective in treatment of short stature associated with short stature homeobox-containing gene deficiency: Two-year results of a randomized, controlled, multicenter trial. J. Clin. Endocrinol. Metab. 2007, 92, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Blum, W.F.; Ross, J.L.; Zimmermann, A.G.; Quigley, C.A.; Child, C.J.; Kalifa, G.; Deal, C.; Drop, S.L.; Rappold, G.; Cutler, G.B., Jr. GH treatment to final height produces similar height gains in patients with SHOX deficiency and Turner syndrome: Results of a multicenter trial. J. Clin. Endocrinol. Metab. 2013, 98, E1383–E1392. [Google Scholar] [CrossRef]
- Shears, D.J.; Guillen-Navarro, E.; Sempere-Miralles, M.; Domingo-Jimenez, R.; Scambler, P.J.; Winter, R.M. Pseudodominant inheritance of Langer mesomelic dysplasia caused by a SHOX homeobox missense mutation. Am. J. Med. Genet. 2002, 110, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Espiritu, C.; Chen, H.; Woolley, P.V., Jr. Mesomelic dwarfism as the homozygous expression of dyschondrosteosis. Am. J. Dis. Child. 1975, 129, 375–377. [Google Scholar] [CrossRef] [PubMed]
- Gleiss, A.; Lassi, M.; Blumel, P.; Borkenstein, M.; Kapelari, K.; Mayer, M.; Schemper, M.; Hausler, G. Austrian height and body proportion references for children aged 4 to under 19 years. Ann. Hum. Biol. 2013, 40, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Flügel, B.; Greil, H.; Sommer, K. Anthropologischer Atlas Grundlagen und Daten. Alters- und Geschlechtsvariabilität des Menschen; Wotzel: Frankfurt/Main, Germany, 1986. [Google Scholar]
- Fredriks, A.M.; van Buuren, S.; van Heel, W.J.; Dijkman-Neerincx, R.H.; Verloove-Vanhorick, S.P.; Wit, J.M. Nationwide age references for sitting height, leg length, and sitting height/height ratio, and their diagnostic value for disproportionate growth disorders. Arch. Dis. Child. 2005, 90, 807–812. [Google Scholar] [CrossRef] [Green Version]
- Gerver, W.J.M.; Gkourogianni, A.; Dauber, A.; Nilsson, O.; Wit, J.M. Arm Span and Its Relation to Height in a 2- to 17-Year-Old Reference Population and Heterozygous Carriers of ACAN Variants. Horm. Res. Paediatr. 2020, 93, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Quanjer, P.H.; Capderou, A.; Mazicioglu, M.M.; Aggarwal, A.N.; Banik, S.D.; Popovic, S.; Tayie, F.A.; Golshan, M.; Ip, M.S.; Zelter, M. All-age relationship between arm span and height in different ethnic groups. Eur. Respir. J. 2014, 44, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Boerkoel, C.F.; Exelbert, R.; Nicastri, C.; Nichols, R.C.; Miller, F.W.; Plotz, P.H.; Raben, N. Leaky splicing mutation in the acid maltase gene is associated with delayed onset of glycogenosis type II. Am. J. Hum. Genet. 1995, 56, 887–897. [Google Scholar]
- Nakagama, Y.; Hamanaka, K.; Mimaki, M.; Shintaku, H.; Miyatake, S.; Matsumoto, N.; Hirohata, K.; Inuzuka, R.; Oka, A. Leaky splicing variant in sepiapterin reductase deficiency: Are milder cases escaping diagnosis? Neurol. Genet. 2019, 5, e319. [Google Scholar] [CrossRef] [Green Version]
- Brejchova, K.; Dudakova, L.; Skalicka, P.; Dobrovolny, R.; Masek, P.; Putzova, M.; Moosajee, M.; Tuft, S.J.; Davidson, A.E.; Liskova, P. IPSC-Derived Corneal Endothelial-like Cells Act as an Appropriate Model System to Assess the Impact of SLC4A11 Variants on Pre-mRNA Splicing. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3084–3090. [Google Scholar] [CrossRef] [Green Version]
- Chin, H.L.; Lin, S.; Dalmann, J.; Modi, B.; Alderman, E.; Salman, A.; Del Bel, K.L.; Lehman, A.; Turvey, S.E.; Boerkoel, C.F. Can leaky splicing and evasion of premature termination codon surveillance contribute to the phenotypic variability in Alkuraya-Kucinskas syndrome? Eur. J. Med. Genet. 2022, 65, 104427. [Google Scholar] [CrossRef]
- Dobrovolny, R.; Liskova, P.; Ledvinova, J.; Poupetova, H.; Asfaw, B.; Filipec, M.; Jirsova, K.; Kraus, J.; Elleder, M. Mucolipidosis IV: Report of a case with ocular restricted phenotype caused by leaky splice mutation. Am. J. Ophthalmol. 2007, 143, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Decker, E.; Durand, C.; Bender, S.; Rodelsperger, C.; Glaser, A.; Hecht, J.; Schneider, K.U.; Rappold, G. FGFR3 is a target of the homeobox transcription factor SHOX in limb development. Hum. Mol. Genet. 2011, 20, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Beiser, K.U.; Glaser, A.; Kleinschmidt, K.; Scholl, I.; Roth, R.; Li, L.; Gretz, N.; Mechtersheimer, G.; Karperien, M.; Marchini, A.; et al. Identification of novel SHOX target genes in the developing limb using a transgenic mouse model. PLoS ONE 2014, 9, e98543. [Google Scholar] [CrossRef] [Green Version]
- Hristov, G.; Marttila, T.; Durand, C.; Niesler, B.; Rappold, G.A.; Marchini, A. SHOX triggers the lysosomal pathway of apoptosis via oxidative stress. Hum. Mol. Genet. 2014, 23, 1619–1630. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, S.; Roeth, R.; Diebold, S.; Gogel, J.; Hassel, D.; Just, S.; Rappold, G.A. Identification and Tissue-Specific Characterization of Novel SHOX-Regulated Genes in Zebrafish Highlights SOX Family Members Among Other Genes. Front. Genet. 2021, 12, 688808. [Google Scholar] [CrossRef]
- Aza-Carmona, M.; Barca-Tierno, V.; Hisado-Oliva, A.; Belinchon, A.; Gorbenko-del Blanco, D.; Rodriguez, J.I.; Benito-Sanz, S.; Campos-Barros, A.; Heath, K.E. NPPB and ACAN, two novel SHOX2 transcription targets implicated in skeletal development. PLoS ONE 2014, 9, e83104. [Google Scholar] [CrossRef] [PubMed]
- McKusick, V.A. Mendelian Inheritance in Man; Johns Hopkins University Press: Baltimore, MD, USA, 1966. [Google Scholar]
- Zschocke, J.; Byers, P.H.; Wilkie, A.O.M. Gregor Mendel and the concepts of dominance and recessiveness. Nat. Rev. Genet. 2022, 23, 387–388. [Google Scholar] [CrossRef]
- Fanelli, A.; Vannelli, S.; Babu, D.; Mellone, S.; Cucci, A.; Monzani, A.; Al Essa, W.; Secco, A.; Follenzi, A.; Bellone, S.; et al. Copy number variations residing outside the SHOX enhancer region are involved in Short Stature and Leri-Weill dyschondrosteosis. Mol. Genet. Genomic. Med. 2022, 10, e1793. [Google Scholar] [CrossRef]
- Vodopiutz, J.; Zoller, H.; Fenwick, A.L.; Arnhold, R.; Schmid, M.; Prayer, D.; Muller, T.; Repa, A.; Pollak, A.; Aufricht, C.; et al. Homozygous SALL1 mutation causes a novel multiple congenital anomaly-mental retardation syndrome. J. Pediatr. 2013, 162, 612–617. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wu, N.; Deciphering Disorders Involving Scoliosis and COmorbidities (DISCO) Study; Yang, N.; Takeda, K.; Chen, W.; Li, W.; Du, R.; Liu, S.; Zhou, Y.; et al. TBX6-associated congenital scoliosis (TACS) as a clinically distinguishable subtype of congenital scoliosis: Further evidence supporting the compound inheritance and TBX6 gene dosage model. Genet. Med. 2019, 21, 1548–1558. [Google Scholar] [CrossRef]
- Lupski, J.R. Clan genomics: From OMIM phenotypic traits to genes and biology. Am. J. Med. Genet. A 2021, 185, 3294–3313. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R. Biology in balance: Human diploid genome integrity, gene dosage, and genomic medicine. Trends Genet. 2022, 38, 554–571. [Google Scholar] [CrossRef] [PubMed]
- Schiller, S.; Spranger, S.; Schechinger, B.; Fukami, M.; Merker, S.; Drop, S.L.; Troger, J.; Knoblauch, H.; Kunze, J.; Seidel, J.; et al. Phenotypic variation and genetic heterogeneity in Leri-Weill syndrome. Eur. J. Hum. Genet. 2000, 8, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalbano, A.; Juergensen, L.; Roeth, R.; Weiss, B.; Fukami, M.; Fricke-Otto, S.; Binder, G.; Ogata, T.; Decker, E.; Nuernberg, G.; et al. Retinoic acid catabolizing enzyme CYP26C1 is a genetic modifier in SHOX deficiency. EMBO Mol. Med. 2016, 8, 1455–1469. [Google Scholar] [CrossRef]
- Montalbano, A.; Juergensen, L.; Fukami, M.; Thiel, C.T.; Hauer, N.H.; Roeth, R.; Weiss, B.; Naiki, Y.; Ogata, T.; Hassel, D.; et al. Functional missense and splicing variants in the retinoic acid catabolizing enzyme CYP26C1 in idiopathic short stature. Eur. J. Hum. Genet. 2018, 26, 1113–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, G.; Renz, A.; Martinez, A.; Keselman, A.; Hesse, V.; Riedl, S.W.; Hausler, G.; Fricke-Otto, S.; Frisch, H.; Heinrich, J.J.; et al. SHOX haploinsufficiency and Leri-Weill dyschondrosteosis: Prevalence and growth failure in relation to mutation, sex, and degree of wrist deformity. J. Clin. Endocrinol. Metab. 2004, 89, 4403–4408. [Google Scholar] [CrossRef] [Green Version]
- Benito-Sanz, S.; Thomas, N.S.; Huber, C.; Gorbenko del Blanco, D.; Aza-Carmona, M.; Crolla, J.A.; Maloney, V.; Rappold, G.; Argente, J.; Campos-Barros, A.; et al. A novel class of Pseudoautosomal region 1 deletions downstream of SHOX is associated with Leri-Weill dyschondrosteosis. Am. J. Hum. Genet. 2005, 77, 533–544. [Google Scholar] [CrossRef] [Green Version]
- Rosilio, M.; Huber-Lequesne, C.; Sapin, H.; Carel, J.C.; Blum, W.F.; Cormier-Daire, V. Genotypes and phenotypes of children with SHOX deficiency in France. J. Clin. Endocrinol. Metab. 2012, 97, E1257–E1265. [Google Scholar] [CrossRef] [Green Version]
- Durand, C.; Roeth, R.; Dweep, H.; Vlatkovic, I.; Decker, E.; Schneider, K.U.; Rappold, G. Alternative splicing and nonsense-mediated RNA decay contribute to the regulation of SHOX expression. PLoS ONE 2011, 6, e18115. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vodopiutz, J.; Steurer, L.-M.; Haufler, F.; Laccone, F.; Garczarczyk-Asim, D.; Hilkenmeier, M.; Steinbauer, P.; Janecke, A.R. Leri–Weill Dyschondrosteosis Caused by a Leaky Homozygous SHOX Splice-Site Variant. Genes 2023, 14, 877. https://doi.org/10.3390/genes14040877
Vodopiutz J, Steurer L-M, Haufler F, Laccone F, Garczarczyk-Asim D, Hilkenmeier M, Steinbauer P, Janecke AR. Leri–Weill Dyschondrosteosis Caused by a Leaky Homozygous SHOX Splice-Site Variant. Genes. 2023; 14(4):877. https://doi.org/10.3390/genes14040877
Chicago/Turabian StyleVodopiutz, Julia, Lisa-Maria Steurer, Florentina Haufler, Franco Laccone, Dorota Garczarczyk-Asim, Matthias Hilkenmeier, Philipp Steinbauer, and Andreas R. Janecke. 2023. "Leri–Weill Dyschondrosteosis Caused by a Leaky Homozygous SHOX Splice-Site Variant" Genes 14, no. 4: 877. https://doi.org/10.3390/genes14040877
APA StyleVodopiutz, J., Steurer, L.-M., Haufler, F., Laccone, F., Garczarczyk-Asim, D., Hilkenmeier, M., Steinbauer, P., & Janecke, A. R. (2023). Leri–Weill Dyschondrosteosis Caused by a Leaky Homozygous SHOX Splice-Site Variant. Genes, 14(4), 877. https://doi.org/10.3390/genes14040877