

SATB2-Associated Syndrome Due to a c.715C>T:p(Arg239*) Variant in Adulthood: Natural History and Literature Review

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Sample
2.2. Whole-Exome Sequencing (WES)
2.3. Data Analysis
2.4. Validation by Sanger Sequencing
3. Case Report
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glass, I.A.; Swindlehurst, C.A.; Aitken, D.A.; McCrea, W.; Boyd, E. Interstitial deletion of the long arm of chromosome 2 with normal levels of isocitrate dehydrogenase. J. Med. Genet. 1989, 26, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, M.; Smith, K.; Basel-Vanagaite, V.L.; Feingold, M.; Brock, P.; Gowans, G.; Vasudevan, P.; Cresswell, L.; Taylor, E.J.; Harris, C.; et al. Case series: 2q33.1 microdeletion syndrome-further delineation of the phenotype. J. Med. Genet. 2011, 48, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Zarate, Y.; Kalsner, L.; Basinger, A.; Jones, J.; Li, C.; Szybowska, M.; Xu, Z.; Vergano, S.; Caffrey, A.; Gonzalez, C.; et al. Genotype and phenotype in 12 additional individuals with SATB2-associated syndrome. Clin. Genet. 2017, 92, 423–429. [Google Scholar] [CrossRef]
- Bengani, H.; Handley, M.; Alvi, M.; Ibitoye, R.; Lees, M.; Lynch, S.A.; Lam, W.; Fannemel, M.; Nordgren, A.; Malmgren, H.; et al. Clinical and molecular consequences of disease-associated de novo mutations in SATB2. Genet. Med. 2017, 19, 900–908. [Google Scholar] [CrossRef] [Green Version]
- Leoyklang, P.; Suphapeetiporn, K.; Siriwan, P.; Desudchit, T.; Chaowanapanja, P.; A Gahl, W.; Shotelersuk, V. Heterozygous nonsense mutation SATB2 associated with cleft palate, osteoporosis, and cognitive defects. Hum. Mutat. 2007, 28, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Döcker, D.; Schubach, M.; Menzel, M.; Munz, M.; Spaich, C.; Biskup, S.; Bartholdi, D. Further delineation of the SATB2 phenotype. Eur. J. Hum. Genet. 2014, 22, 1034–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Buggenhout, G.; Van Ravenswaaij-Arts, C.; MC Maas, N.; Thoelen, R.; Vogels, A.; Smeets, D.; Salden, I.; Matthijs, G.; Fryns, J.-P.; Vermeesch, J. The del(2)(q32.2q33) deletion syndrome defined by clinical and molecular characterization of four patients. Eur. J. Med. Genet. 2005, 48, 276–289. [Google Scholar] [CrossRef]
- Mencarelli, M.A.; Caselli, R.; Pescucci, C.; Hayek, J.; Zappella, M.; Renieri, A.; Mari, F. Clinical and molecular characterization of a patient with a 2q31.2-32.3 deletion identified by array-CGH. Am. J. Med. Genet. Part A 2007, 143A, 858–865. [Google Scholar] [CrossRef]
- de Ravel, T.J.; Balikova, I.; Thiry, P.; Vermeesch, J.R.; Frijns, J.-P. Another patient with a de novo deletion further delineates the 2q33.1 microdeletion syndrome. Eur. J. Med. Genet. 2009, 52, 120–122. [Google Scholar] [CrossRef]
- Rosenfeld, J.A.; Ballif, B.C.; Lucas, A.; Spence, E.J.; Powell, C.; Aylsworth, A.S.; Torchia, B.A.; Shaffer, L.G. Small deletions of SATB2 cause some of the clinical features of the 2q33.1 microdeletion syndrome. PLoS ONE 2009, 4, e6568. [Google Scholar] [CrossRef] [Green Version]
- Urquhart, J.; Black, G.C.; Clayton-Smith, J. 4.5 Mb microdeletion in chromosome band 2q33.1 associated with learning disability and cleft palate. Eur. J. Med. Genet. 2009, 52, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Cocchella, A.; Malacarne, M.; Forzano, F.; Marciano, C.; Pierluigi, M.; Perroni, L.; Faravelli, F.; Di Maria, E. The refinement of the critical region for the 2q31.2q32.3 deletion syndrome indicates candidate genes for mental retardation and speech impairment. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.I.; Matoso, E.; Venancio, M.; Saraiva, J.; Melo, J.B.; Carreira, I.M. Critical region in 2q31.2q32.3 deletion syndrome: Report of two phenotypically distinct patients, one with an additional deletion in Alagille syndrome region. Mol. Cytogenet. 2012, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaiser, A.-S.; Maas, B.; Wolff, A.; Sutter, C.; Janssen, J.W.; Hinderhofer, K.; Moog, U. Characterization of the first intragenic SATB2 duplication in a girl with intellectual disability, nearly absent speech and suspected hypodontia. Eur. J. Med. Genet. 2015, 23, 704–707. [Google Scholar] [CrossRef] [Green Version]
- Zarate, Y.A.; Perry, H.; Ben-Omran, T.; Sellars, E.A.; Stein, Q.; Almureikhi, M.; Simmons, K.; Klein, O.; Fish, J.; Feingold, M.; et al. Further supporting evidence for the SATB2-associated syndrome found through whole exome sequencing. Am. J. Med. Genet. Part A 2015, 167A, 1026–1032. [Google Scholar] [CrossRef]
- Zarate, Y.A.; Bosanko, K.A.; Caffrey, A.R.; Bernstein, J.A.; Martin, D.M.; Williams, M.S.; Berry-Kravis, E.M.; Mark, P.R.; Manning, M.A.; Bhambhani, V.; et al. Mutation update for the SATB2 gene. Hum. Mutat. 2019, 40, 1013–1029. [Google Scholar] [CrossRef] [Green Version]
- FitzPatrick, D.R.; Carr, I.M.; McLaren, L.; Leek, J.P.; Wightman, P.; Williamson, K.; Gautier, P.; I McGill, N.; Hayward, C.; Firth, H.V.; et al. Identification of SATB2 as the cleft palate gene on 2q32-q33. Hum. Mol. Genet. 2003, 12, 2491–2501. [Google Scholar] [CrossRef] [Green Version]
- Dobreva, G.; Dambacher, J.; Grosschedl, R. SUMO modification of a novel MAR-binding protein, SATB2, modulates immunoglobulin mu gene expression. Genes Dev. 2003, 17, 3048–3061. [Google Scholar] [CrossRef] [Green Version]
- Szemes, M.; Gyorgy, A.; Paweletz, C.; Dobi, A.; Agoston, D.V. Isolation and characterization of SATB2, a novel AT-rich DNA binding protein expressed in development and cell-specific manner in the rat brain. Neurochem. Res. 2006, 31, 237–246. [Google Scholar] [CrossRef]
- Dobreva, G.; Chahrour, M.; Dautzenberg, M.; Chirivella, L.; Kanzler, B.; Fariñas, I.; Karsenty, G.; Grosschedl, R. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell 2006, 125, 971–986. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Song, N.-N.; Lan, W.; Hu, L.; Su, C.-J.; Ding, Y.-Q.; Zhang, L. Expression of transcription factor Satb2 in adult mouse brain. Anat. Rec. 2013, 296, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Britanova, O.; Depew, M.J.; Schwark, M.; Thomas, B.L.; Miletich, I.; Sharpe, P.; Tarabykin, V. Satb2 haploinsufficiency phenocopies 2q32-q33 deletions, whereas loss suggests a fundamental role in the coordination of jaw development. Am. J. Hum. Genet. 2006, 79, 668–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liedén, A.; Kvarnung, M.; Nilssson, D.; Sahlin, E.; Lundberg, E.S. Intragenic duplication—A novel causative mechanism for SATB2-associated syndrome. Am. J. Med. Genet. Part A 2014, 164A, 3083–3087. [Google Scholar] [CrossRef] [PubMed]
- Leoyklang, P.; Suphapeetiporn, K.; Srichomthong, C.; Tongkobpetch, S.; Fietze, S.; Dorward, H.; Cullinane, A.R.; Gahl, W.A.; Huizing, M.; Shotelersuk, V. Disorders with similar clinical phenotypes reveal underlying genetic interaction: SATB2 acts as an activator of the UPF3B gene. Hum. Genet. 2013, 132, 1383–1393. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Uehara, T.; Suzuki, H.; Takenouchi, T.; Yoshihashi, H.; Suzumura, H.; Mizuno, S.; Kosaki, K. SATB2-associated syndrome in patients from Japan: Linguistic profiles. Am. J. Med. Genet. Part A 2019, 179, 896–899. [Google Scholar] [CrossRef] [Green Version]
- Dowrey, T.; Schwager, E.E.; Duong, J.; Merkuri, F.; Zarate, Y.A.; Fish, J.L. Satb2 regulates proliferation and nuclear integrity of pre-osteoblasts. Bone 2019, 127, 488–498. [Google Scholar] [CrossRef]
- Mouillé, M.; Rio, M.; Breton, S.; Piketty, M.L.; Afenjar, A.; Amiel, J.; Capri, Y.; Goldenberg, A.; Francannet, C.; Michot, C.; et al. SATB2-associated syndrome: Characterization of skeletal features and of bone fragility in a prospective cohort of 19 patients. Orphanet J. Rare Dis. 2022, 17, 100. [Google Scholar] [CrossRef]
- Zarate, Y.A.; Kannan, A.; Bosanko, K.A.; Caffrey, A.R. Growth in individuals with SATB2-associated syndrome. Am. J. Med. Genet. Part A 2022, 188, 2952–2957. [Google Scholar] [CrossRef]
- Zarate, Y.A.; Smith-Hicks, C.L.; Greene, C.; Abbott, M.; Siu, V.M.; Calhoun, A.R.U.L.; Pandya, A.; Li, C.; Sellars, E.A.; Kaylor, J.; et al. Natural history and genotype-phenotype correlations in 72 individuals with SATB2-associated syndrome. Am. J. Med. Genet. Part A 2018, 176A, 925–935. [Google Scholar] [CrossRef] [Green Version]
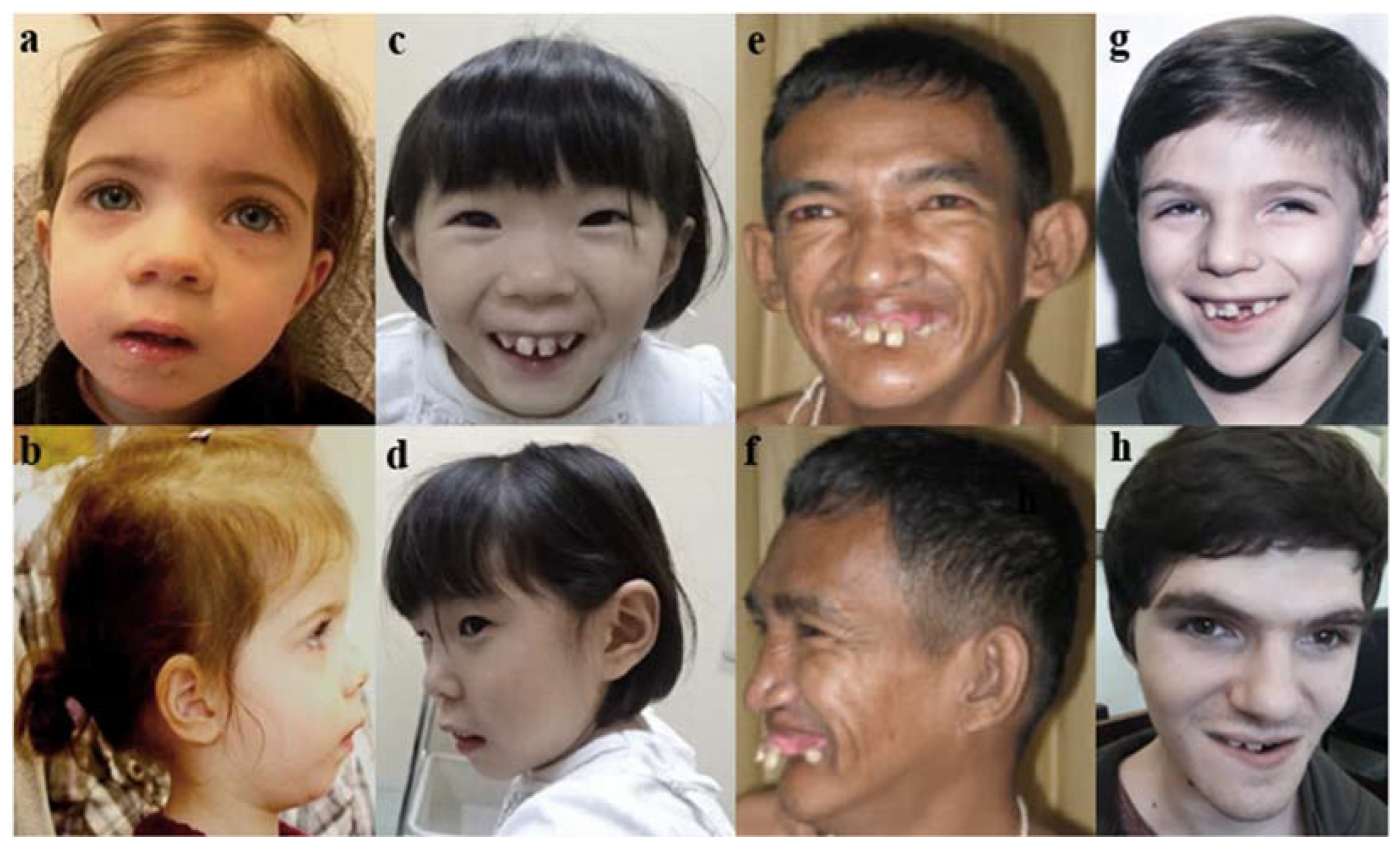

{kind=link}
| Study | Present Study | [5] | [6] | [29] Case 33 | [29] Case 76 | [16] Case 114 | [16] Case 121 | [16] Case 139 | [25] | All Cases with c.715 C>T Variant | All Cases Reported † |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Origin | de novo | de novo | de novo | unknown | unknown | de novo | unknown | unknown | de novo | 100% de novo (5/5) | 96.7% de novo (120/124) |
| Age at Time of Study (years) | 25 | 36 | 3 | 6.5 | 3 | 5 | 9.5 | 3 | 7 | 10.8 | 11 |
| Gender | male | male | female | female | male | male | male | male | female | male 66.6% (6/9) | male 56.6% (86/152) |
| Absence of Speech | + | one word | + | + | one word | one word | one word | + | + | 55.5% (5/9) | 43.8% (64/146) |
| DD/ID | + | + | + | + | + | + | + | + | + | 100% (9/9) | 100% (151/151) |
| Cleft Palate | + | + | + | + | + | − | + | + | + | 88.8% (8/9) | 43.9% (65/148) |
| Feeding Difficulties | − | + | − | + | + | + | + | + | − | 66.6% (6/9) | 66.9% (83/124) |
| Dental Abnormalities | + | + | + | + | + | + | + | + | + | 100% (9/9) | 98.4% (129/131) |
| Dysmorphic Facial Features | + | + | + | + | + | + | + | + | + | 100% (9/9) | 83.4% (101/121) |
| Low Bone Mineral Density | + | + | − | + | − | NR | − | NR | − | 42.8% (3/7) | 70.4% (31/44) |
| Congenital Heart Disease | + | − | − | − | − | − | − | − | − | 11.1% (1/9) | 0% (0/152) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Copelli, M.d.M.; Pairet, E.; Atique-Tacla, M.; Vieira, T.P.; Appenzeller, S.; Helaers, R.; Vikkula, M.; Gil-da-Silva-Lopes, V.L. SATB2-Associated Syndrome Due to a c.715C>T:p(Arg239*) Variant in Adulthood: Natural History and Literature Review. Genes 2023, 14, 882. https://doi.org/10.3390/genes14040882
Copelli MdM, Pairet E, Atique-Tacla M, Vieira TP, Appenzeller S, Helaers R, Vikkula M, Gil-da-Silva-Lopes VL. SATB2-Associated Syndrome Due to a c.715C>T:p(Arg239*) Variant in Adulthood: Natural History and Literature Review. Genes. 2023; 14(4):882. https://doi.org/10.3390/genes14040882
Chicago/Turabian StyleCopelli, Matheus de Mello, Eleonore Pairet, Milena Atique-Tacla, Társis Paiva Vieira, Simone Appenzeller, Raphaël Helaers, Miikka Vikkula, and Vera Lúcia Gil-da-Silva-Lopes. 2023. "SATB2-Associated Syndrome Due to a c.715C>T:p(Arg239*) Variant in Adulthood: Natural History and Literature Review" Genes 14, no. 4: 882. https://doi.org/10.3390/genes14040882
APA StyleCopelli, M. d. M., Pairet, E., Atique-Tacla, M., Vieira, T. P., Appenzeller, S., Helaers, R., Vikkula, M., & Gil-da-Silva-Lopes, V. L. (2023). SATB2-Associated Syndrome Due to a c.715C>T:p(Arg239*) Variant in Adulthood: Natural History and Literature Review. Genes, 14(4), 882. https://doi.org/10.3390/genes14040882