
Comprehensive Genetic Analysis of Druze Provides Insights into Carrier Screening
 , , , and
, , , and
Abstract
1. Introduction
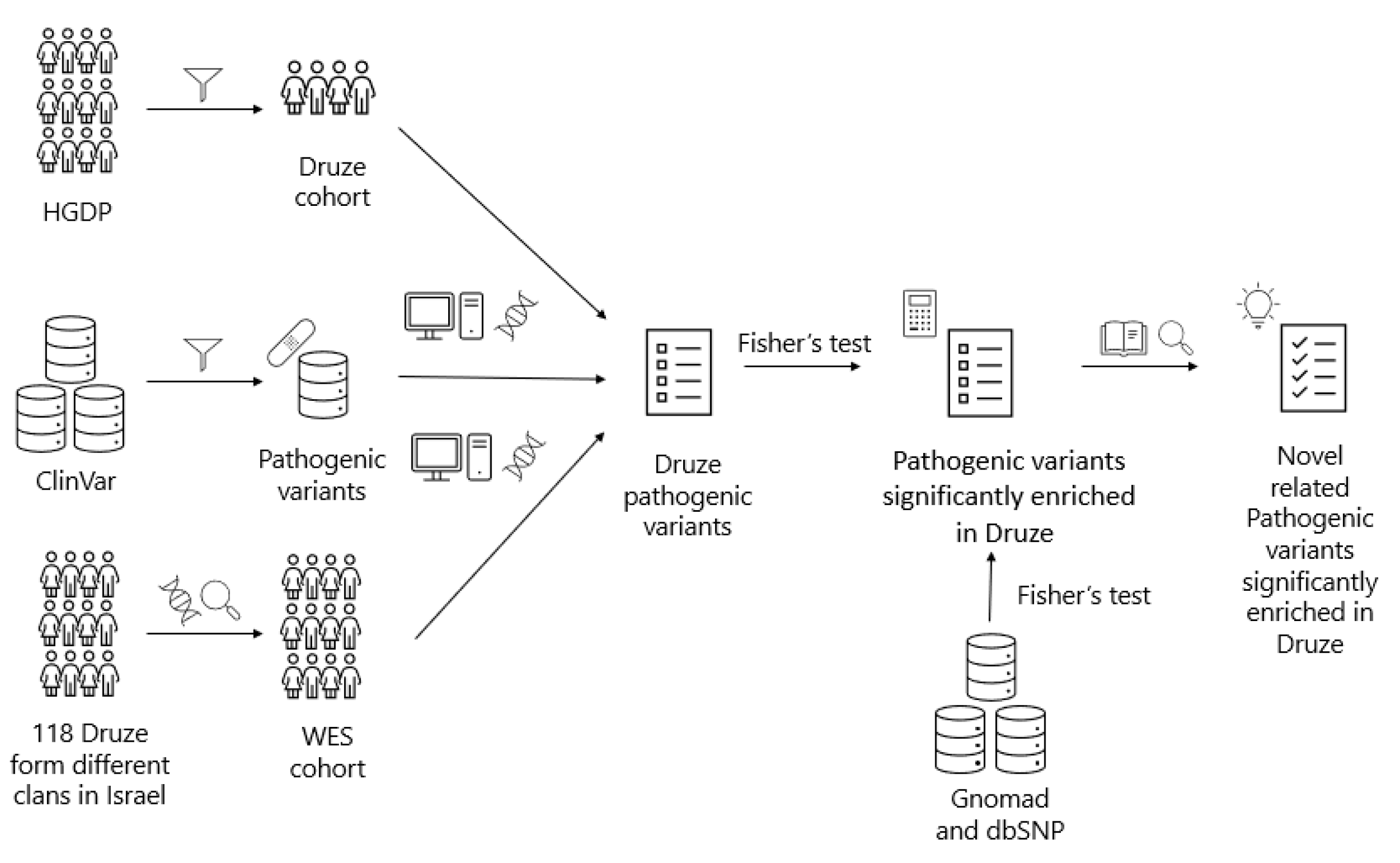
2. Materials and Methods
Recruitment of Druze Participants for WES
3. Results
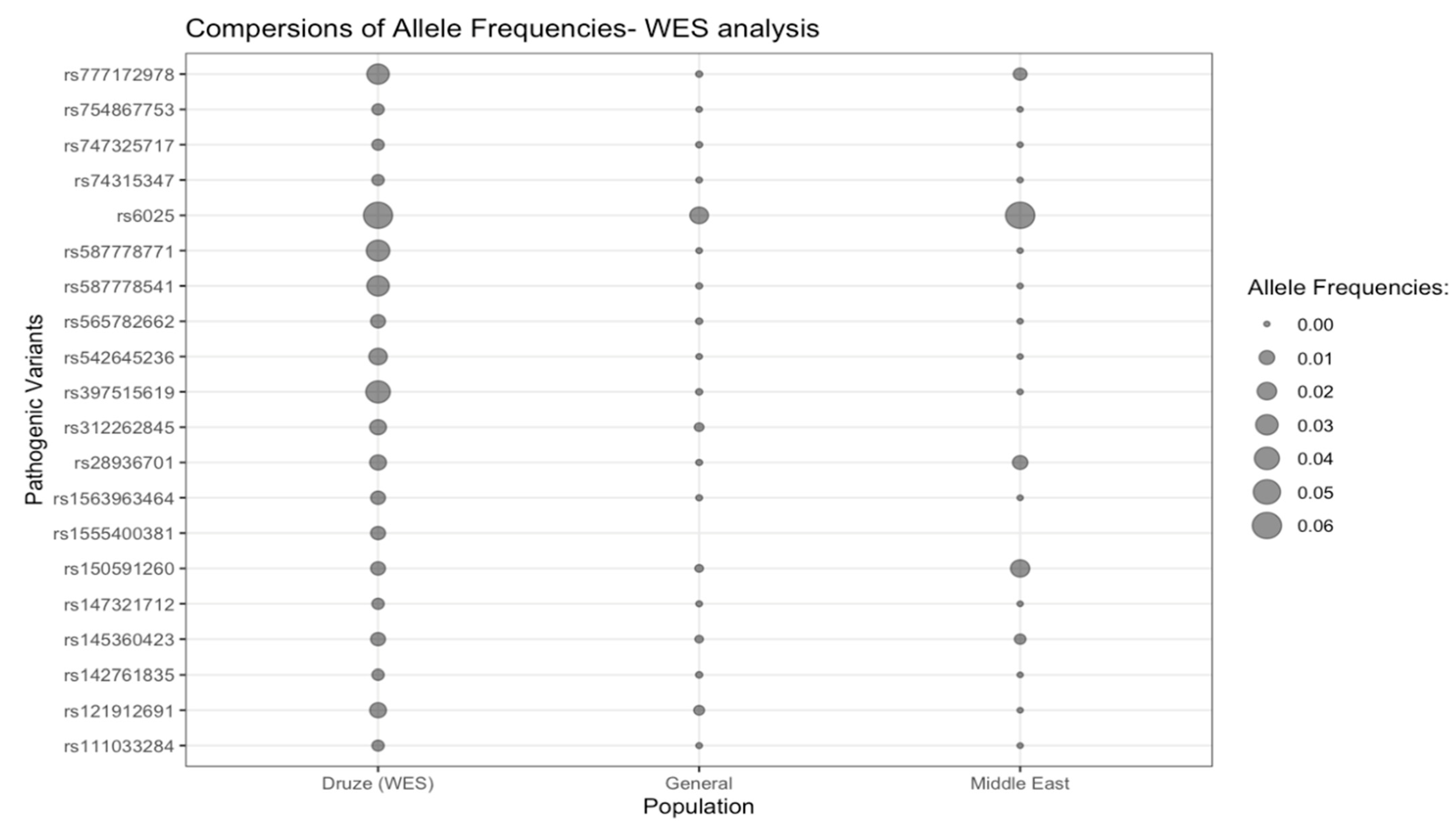
3.1. WES Analysis
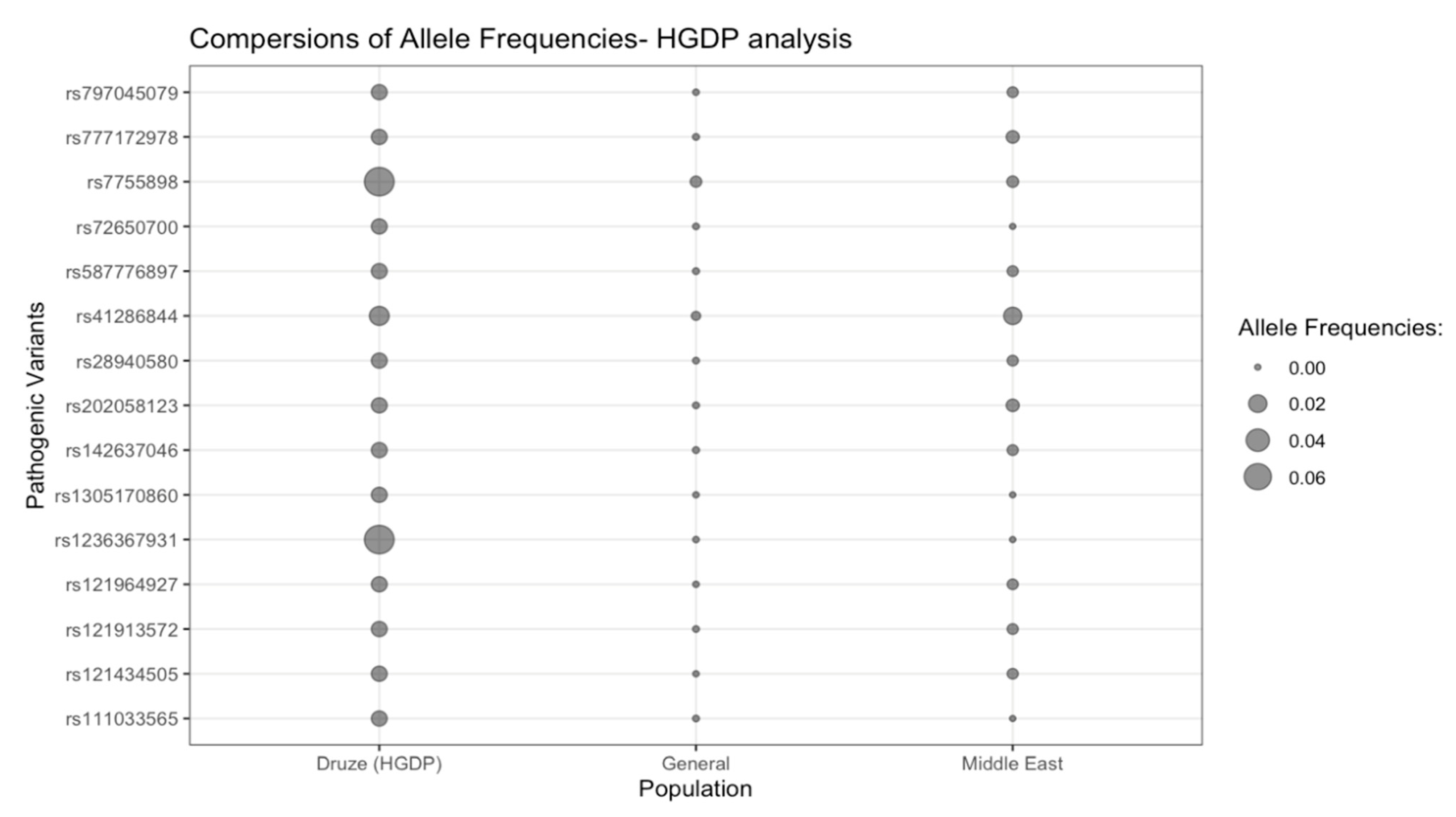
3.2. HGDP Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Swayd, S. The Druzes: An Annotated Bibliography; ISES Publications: Kirkland, WA, USA, 1998. [Google Scholar]
- Miles, W.F.S. As the Druze Go, So Goes the Middle East. Curr. Hist. 2021, 120, 366–371. [Google Scholar] [CrossRef]
- Vardi-Saliternik, R.; Friedlander, Y.; Cohen, T. Consanguinity in a population sample of Israeli Muslim Arabs, Christian Arabs and Druze. Ann. Hum. Biol. 2002, 29, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Fares, F.; Ran, S.A.; David, M.; Zelnik, N.; Hecht, Y.; Khairaldeen, H.; Lerner, A. Identification of two mutations for ataxia telangiectasia among the Druze community. Prenat. Diagn. 2004, 24, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Filon, D.; Oron, V.; Krichevski, S.; Shaag, A.; Shaag, Y.; Warren, T.C.; Goldfarb, A.; Shneor, Y.; Koren, A.; Aker, M. Diversity of beta-globin mutations in Israeli ethnic groups reflects recent historic events. Am. J. Hum. Genet. 1994, 54, 836–843. [Google Scholar] [PubMed]
- Landsberger, D.; Meiner, V.; Reshef, A.; Levy, Y.; Van Der Westhuyzen, D.R.; Coetzee, A.G.; Leitersdorf, E. A nonsense mutation in the LDL receptor gene leads to familial hypercholesterolemia in the Druze sect. Am. J. Hum. Genet. 1992, 50, 427–433. [Google Scholar]
- Zlotogora, J. Autosomal recessive diseases among the Israeli Arabs. Hum. Genet. 2019, 138, 1117–1122. [Google Scholar] [CrossRef]
- Haber, M.; Gauguier, D.; Youhanna, S.; Patterson, N.; Moorjani, P.; Botigué, L.R.; Platt, D.E.; Matisoo-Smith, E.; Soria-Hernanz, D.F.; Wells, R.S.; et al. Genome-Wide Diversity in the Levant Reveals Recent Structuring by Culture. PLoS Genet. 2013, 9, e1003316. [Google Scholar] [CrossRef]
- Zidan, J.; Ben-Avraham, D.; Carmi, S.; Maray, T.; Friedman, E.; Gil Atzmon, G. Genotyping of geographically diverse Druze trios reveals substructure and a recent bottleneck. Eur. J. Hum. Genet. 2015, 23, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Bergström, A.; McCarthy, S.A.; Hui, R.; Almarri, M.A.; Ayub, Q.; Danecek, P.; Chen, Y.; Felkel, S.; Hallast, P.; Kamm, J.; et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 2020, 367, eaay5012. [Google Scholar] [CrossRef]
- Strauss, K.A.; Gonzaga-Jauregui, C.; Brigatti, K.W.; Williams, K.B.; King, A.K.; Van Hout, C.; Robinson, D.L.; Young, M.; Praveen, K.; Heaps, A.D.; et al. Genomic diagnostics within a medically underserved population: Efficacy and implications. Genet. Med. 2018, 20, 31–41. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Khayat, M.; Danial-Farran, N.; Chervinsky, E.; Zehavi, Y.; Peled-Peretz, L.; Gafni-Amsalem, C.; Hakrosh, S.; Abu-Leil Zouabi, O.; Tamir, L.; Mamlouk, E.; et al. The landscape of autosomal recessive variants in an isolated community: Implications for population screening for reproductive purposes. Clin. Genet. 2021, 100, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.T.; Maloney, K.A.; Pollin, T.I.; Streeten, E.A.; Xu, H.; Shuldiner, A.R.; Van Hout, C.V.; Gonzaga-Jauregui, C.; Mitchell, B.D.; Center, R.G. The burden of pathogenic variants in clinically actionable genes in a founder population. Am. J. Med. Genet. Part A 2021, 185, 3476–3484. [Google Scholar] [CrossRef]
- Zlotogora, J. The Israeli national population program of genetic carrier screening for reproductive purposes. How should it be continued? Isr. J. Health Policy Res. 2019, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Amiel, A.; Hijazi, S.; Ezra, D.; Tentler, Y.; Halabi, S. Compliance of Israeli Druze Women to Undergo a Non-Invasive Prenatal Test. Asian Res. J. Gynaecol. Obstet. 2021, 6, 9–16. [Google Scholar]
- Machicado, J.D.; Yadav, D. Epidemiology of Recurrent Acute and Chronic Pancreatitis: Similarities and Differences. Dig. Dis. Sci. 2017, 62, 1683–1691. [Google Scholar] [CrossRef]
- Balagura, G.; Riva, A.; Marchese, F.; Iacomino, M.; Madia, F.; Giacomini, T.; Mancardi, M.M.; Amadori, E.; Vari, M.S.; Salpietro, V.; et al. Clinical spectrum and genotype-phenotype correlations in PRRT2 Italian patients. Eur. J. Paediatr. Neurol. 2020, 28, 193–197. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; Bale, S.J.; et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1381–1390. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Ogata, H.; Katsumata, D.; Nakajima, M.; Fujii, T.; Tsutsumi, S.; Asao, T.; Sasaki, K.; Kuwano, H.; Kato, H. MUTYH-associated colorectal cancer and adenomatous polyposis. Surg. Today 2014, 44, 593–600. [Google Scholar] [CrossRef]
- Manna, R.; Rigante, D. Familial Mediterranean Fever: Assessing the Overall Clinical Impact and Formulating Treatment Plans. Mediterr. J. Hematolol. Infect. Dis. 2019, 11, e2019027. [Google Scholar]
- Woodage, T.; King, S.M.; Wacholder, S.; Hartge, P.; Struewing, J.P.; McAdams, M.; Laken, S.J.; Tucker, M.A.; Brody, L.C. The APCI1307K allele and cancer risk in a community-based study of Ashkenazi Jews. Nat. Genet. 1998, 20, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Laken, S.J.; Petersen, G.M.; Gruber, S.B.; Oddoux, C.; Ostrer, H.; Giardiello, F.M.; Hamilton, S.R.; Hampel, H.; Markowitz, A.; Klimstra, D.; et al. Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC. Nat. Genet. 1997, 17, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Rozen, P.; Shomrat, R.; Strul, H.; Naiman, T.; Karminsky∥, N.; Legum, C.; Orr-Urtreger, A. Prevalence of the I1307K APC gene variant in Israeli Jews of differing ethnic origin and risk for colorectal cancer. Gastroenterology 1999, 116, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Chen-Shtoyerman, R.; Theodor, L.; Harmati, E.; Friedman, E.; Dacka, S.; Kopelman, Y.; Sternberg, A.; Zarivach, R.; Bar-Meir, S.; Fireman, Z. Genetic analysis of familial colorectal cancer in Israeli Arabs. Hum. Mutat. 2003, 21, 446–447. [Google Scholar] [CrossRef]
- Liang, J.; Lin, C.; Hu, F.; Wang, F.; Zhu, L.; Yao, X.; Wang, Y.; Zhao, Y. APC polymorphisms and the risk of colorectal neoplasia: A HuGE review and meta-analysis. Am. J. Epidemiol. 2013, 177, 1169–1179. [Google Scholar] [CrossRef]
- Figer, A.; Shtoyerman-Chen, R.; Tamir, A.; Geva, R.; Irmin, L.; Flex, D.; Theodor, L.; Sulkes, A.; Sadetzki, S.; Bar-Meir, S.; et al. Phenotypic characteristics of colo-rectal cancer in I1307K APC germline mutation carriers compared with sporadic cases. Br. J. Cancer 2001, 85, 1368–1371. [Google Scholar] [CrossRef]
- Kiafar, M.; Faezi, S.T.; Kasaeian, A.; Baghdadi, A.; Kakaei, S.; Mousavi, S.A.; Nejadhosseinian, M.; Shahram, F.; Ghodsi, S.Z.; Shams, H.; et al. Diagnosis of Behçet’s disease: Clinical characteristics, diagnostic criteria, and differential diagnoses. BMC Rheumatolplgy 2021, 5, 2. [Google Scholar] [CrossRef]
- Chen, J.; Yao, X. A Contemporary Review of Behcet’s Syndrome. Clin. Rev. Allergy Immunol. 2021, 61, 363–376. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Aslani, S.; Meguro, A.; Akhtari, M.; Fatahi, Y.; Mizuki, N.; Shahram, F. A comprehensive overview on the genetics of Behçet’s disease. Int. Rev. Immunol. 2022, 41, 84–106. [Google Scholar] [CrossRef]
- Dixon, K.O.; Kuchroo, V.K. IL-18: Throwing off the shackles to boost anti-tumor immunity. Cell Res. 2020, 30, 831–832. [Google Scholar] [CrossRef]
- Mesquida, M.; Molins, B.; Llorenç, V.; Hernández, M.V.; Espinosa, G.; Dick, A.D.; Adán, A. Current and future treatments for Behçet’s uveitis: Road to remission. Int. Ophthalmol. 2013, 34, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Pay, S.; Erdem, H.; Pekel, A.; Simsek, I.; Musabak, U.; Sengul, A.; Dinc, A. Synovial proinflammatory cytokines and their correlation with matrix metalloproteinase-3 expression in Behçet’s disease. Does interleukin-1β play a major role in Behçet’s synovitis? Rheumatol. Int. 2015, 26, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Zhou, Q.; Lv, M.; Tan, H.; Wang, Q.; Zhang, L.; Cao, Q.; Yuan, G.; Su, G.; Kijlstra, A.; et al. Functional Genetic Polymorphisms in the IL1RL1–IL18R1 Region Confer Risk for Ocular Behçet’s Disease in a Chinese Han Population. Front. Genet. 2020, 11, 645. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Rs Number | ClinVar ID a | Condition b | Gene (OMIM #) | Nucleotide Alteration (Amino Acid) | Mutation Type | Location c | Durze AF (AC/AN) | Heterozygous/Homozygous | General Population AF (AC/AN) | Fisher’s p-Value | Middle East AF (AC/AN) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs142761835 | 167199 (2, P) | Isovaleryl-CoA dehydrogenase deficiency (AR) | IVD (607036) | NM_002225.5:c.358G > A (NP_002216.3:p.Gly120Arg) | Missense | 15-40410699 G > A | 0.004 (1/236) | 1/0 | 0.0001 (17/152,182) | 0.03 | 0 (0/316) |
| rs747325717 | 916358 (1, LP) | Charcot-Marie-Tooth disease axonal, type 2F (AD) | HSPB1 (602195) | NM_001540.5:c.438dup (NP_001531.1:p.Gly147fs) | Frameshift | 7-76303987 G > GC | 0.004 (1/236) | 1/0 | 7 × 10−5 (11/152,154) | 0.02 | 0 (0/316) |
| rs147321712 | 65804 (2, P) | OTOF-Related Deafness (AR) | OTOF (603681) | NM_194248.3:c.4483C > T (NP_919224.1:p.Arg1495Ter) | Nonsense (stop gain) | 2-26466731 G > A | 0.004 (1/236) | 1/0 | 2 × 10−5 (3/152,210) | 0.006 | 0 (0/316) |
| rs74315347 | 5368 (2, P) | Nephrotic syndrome, type 2 (AR) | NPHS2 (604766) | NC_000001.11:g.179557227C > T (NP_055440.1:p.Val180Met) | Missense | 1-179557227 C > T | 0.004 (1/236) | 1/0 | 1 × 10−5 (2/152,126) | 0.005 | 0 (0/316) |
| rs111033284 | 43340 (2, P) | Inborn genetic diseases (AR) | MYO7A (276903) | NM_000260.4:c.722G > A (NP_000251.3:p.Arg241His) | Missense | 11-77156991 G > A | 0.004 (1/236) | 1/0 | 1 × 10−5 (2/152,244) | 0.005 | 0 (0/316) |
| rs754867753 | 216118 (2, P) | Primary ciliary dyskinesia (AR) | CCDC40 (613799) | NM_017950.4:c.961C > T (NP_060420.2:p.Arg321Ter) | Nonsense (stop gain) | 17-80050085 C > T | 0.004 (1/236) | 1/0 | 7 × 10−6 (1/151,790) | 0.003 | 0 (0/316) |
| rs1555400381 | 558720 (2, P) | Propionic acidemia (AR) | PCCA (232000) | NM_000282.4:c.843del (NP_000273.2:p.Asn281fs) | Inframe deletion | 13-100920966 AT > A | 0.008 (2/236) | 2/0 | NA | NA | NA |
| rs150591260 | 203805 (2, P) | 3-methylcrotonyl CoA carboxylase 2 deficiency (AR) | MCCC2 (609014) | NM_022132.5: c.1015G > A (NP_071415.1:p.Val339Met) | Missense | 5-71641018 G > A | 0.008 (2/236) | 2/0 | 0.0006 (95/152,168) | 0.01 | 0.02 (6/312) |
| rs145360423 | 426990 (2, P) | Chronic granulomatous disease, cytochrome b-positive, type 1 (AR) | NCF1 (608512) | NM_000265.7:c.579G > A (NP_000256.4:p.Trp193Ter) | Nonsense (stop gain) | 7-74783529 G > A | 0.008 (2/236) | 2/0 | 0.0006 (84/151,994) | 0.008 | 0.003 (1/316) |
| rs565782662 d | 5268 (2, P) | Netherton syndrome (AR) | SPINK5 (605010) | NM_006846.3:c.2468dupA (NP_006837.2:p.Lys824fs) | Frameshift | 5-148120311 G > GA | 0.008 (2/236) | 2/0 | 0.0001 (15/146,014) | 0.0003 | 0 (0/312) |
| rs1563963464 d | 634641 (1, P) | Ataxia-oculomotor apraxia type 1 (AR) | APTX (606350) | NC_000009.12:g.32986031C > A | Splice acceptor | 9-32986031 C > A | 0.008 (2/236) | 2/0 | 0.00004 (3/82,310) | 8 × 10−5 | 0 (0/118) |
| rs121912691 | 18115 (2, P) | Cystinuria (AR) | SLC3A1 (104614) | NM_000341.4:c.1400T > C (NP_000332.2:p.Met467Thr) | Missense | 2-44312653 T > C | 0.01 (3/236) | 3/0 | 0.002 (358/152,198 | 0.02 | 0 (0/316) |
| rs312262845 d | 41142 (1, P) | Orofaciodigital syndrome I (XLD) | OFD1 (311200) | NM_003611.3:c.710del (NP_003602.1:p.Lys237fs) | Frameshift | X-13746826 CA > C | 0.01 (3/236) | 3/0 | 0.001 (111/85,096) | 0.004 | NA |
| rs28936701 | 7733 (2, P) | Glaucoma 3A (AR) | CYP1B1 (601771) | NC_000002.12:g.38070949G > A | Missense | 2-38070949 G > A | 0.01 (3/236) | 3/0 | 5.3 × 10−5 (8/152,118) | 6 × 10−7 | 0.01 (3/316) |
| rs542645236 | 225134 (3, P) | Phenylketonuria (AR) | PAH (612349) | NM_000277.3:c.320A > G (NP_000268.1:p.His107Arg) | Missense | 12-102894767 T > C | 0.02 (4/236) | 4/0 | 1 × 10−5 (2/152,204) | 8.4 × 10−11 | 0 (0/316) |
| rs777172978 | 1029383 (1, P) | Ullrich congenital muscular dystrophy, type 1 (AR) | COL6A2 (120240) | NM_058174.3:c.2554C > T (NP_478054.2:p.Gln852Ter) | Nonsense (stop gain) | 21-46129288 C > T | 0.03 (7/236) | 7/0 | 7.2 × 10−5 (11/152,254) | 6.10 × 10−16 | 0.006 (2/316) |
| rs587778541 | 127838 (2, P) | Hereditary cancer-predisposing syndrome (AR) | MUTYH (604933) | NM_001048174.2:c.1350GGA [1] (NP_001041639.1:p.Glu452del) | Inframe deletion | 1-45331218 TTCC > T | 0.03 (7/236) | 7/0 | 5 × 10−5 (8/152,112) | 1.2 × 10−16 | 0 (0/316) |
| rs587778771 d | 39752 (2, LP) | Episodic kinesigenic dyskinesia, type 1 (AR) | PRRT2 (614386) | NM_145239.3:c.649del (NP_660282.2:p.Arg217fs) | Frameshift | 16-29813694 GC > G | 0.03 (8/236) | 8/0 | 1 × 10−5 (2/149,480) | 1.5 × 10−21 | 0 (0/310) |
| rs397515619 | 6306 (1, P) | Spermatogenic failure, type 5 (AR) | AURKC (603495) | NM_001015878.2: c.145del (NP_001015878.1:p.Leu49fs) | Frameshift | 19-57232069 TC > T | 0.04 (9/236) | 9/0 | 0.0001 (14/151,932) | 3.6 × 10−20 | 0 (0/316) |
| rs6025 | 642 (2, P) | Thrombophilia due to factor V Leiden (AD) | F5 (612309) | NM_000130.5:c.1601G > A (NP_000121.2:p.Arg534Gln) | Missense | 1-169549811 C > T | 0.06 (14/236) | 14/0 | 0.02 (2638/152,200) | 8 × 10−5 | 0.06 (19/316) |
| Rs Number | ClinVar ID a | Condition b | Gene (OMIM #) | Nucleotide Alteration (Amino Acid) | Mutation Type | Location c | Durze AF (AC/AN) | Heterozygous/Homozygous | General Population AF (AC/AN) | Fisher’s p-Value | Middle East AF (AC/AN) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| rs1305170860 | 95704 (2, P) | Cobalamin C disease (AR) | MMACHC (609831) | NM_015506.3:c.547_548del (NP_056321.2:p.Val183fs) | Frameshift | 1-45508909 CTG > C | 0.01 (1/80) | 1/0 | 6.6 × 10−6 (1/152,210) | 0.001 | 0 (0/316) |
| rs121964927 | 265135 (2, P) | Congenital factor VII deficiency (AR) | F7 (613878) | NM_019616.4:c.1043G > T (NP_062562.1:p.Cys348Phe) | Missense | 13-113118716 G > T | 0.01 (1/80) | 1/0 | 1.3 × 10−5 (2/152,168) | 0.001 | 0.003 (1/316) |
| rs72650700 | 30339 (2, P) | Pseudoxanthoma Elasticum (AR) | ABCC6 (603234) | NM_001171.6:c.1552C > T (NP_001162.5:p.Arg518Ter) | Nonsense (stop gain) | 16-16190247 G > A | 0.01 (1/80) | 1/0 | 4.6 × 10−5 (7/152,070) | 0.004 | 0 (0/316) |
| rs28940580 | 36507 (2, P) | Familial Mediterranean Fever (AR) | MEFV (608107) | NM_000243.3:c.2040G > C (NP_000234.1: p.Met680Ile) | Missense | 16-3243447 G > C | 0.01 (1/80) | 1/0 | 4.6 × 10−5 (7/152,148) | 0.004 | 0.003 (1/316) |
| rs587776897 | 31015 (2, P) | Desbuquois dysplasia 1 (AR) | CANT1 (613165) | NM_001159773.2:c.277_278del (NP_001153245.1:p.Leu93fs) | Frameshift | 17-78997344 CAG > C | 0.01 (1/80) | 1/0 | 6.6 × 10−5 (10/151,918) | 0.006 | 0.003 (1/316) |
| rs797045079 | 208559 (2, LP) | Renal dysplasia (AR) | ACE (106180) | NM_000789.4:c.12_31del (NP_000780.1:p.Ser5fs) | Frameshift | 17-63477105 CCTCGGGCCGCCGGGGGCCGG > C | 0.01 (1/80) | 1/0 | 2 × 10−5 (3/151,282) | 0.002 | 0.003 (1/312) |
| rs777172978 | 1029383 (1, P) | Ullrich congenital muscular dystrophy 1 (AR) | COL6A2 (120240) | NM_058174.3:c.2554C > T (NP_478054.2:p.Gln852Ter) | Nonsense (stop gain) | 21-46129288 C > T | 0.01 (1/80) | 1/0 | 7.2 × 10−5 (11/152,254) | 0.006 | 0.006 (2/316) |
| rs121913572 | 14296 (2, P) | Merosin-deficient congenital muscular dystrophy (AR) | LAMA2 (156225) | NM_000426.4: c.7732C > T (NP_000417.3:p.Arg2578Ter) | Nonsense (stop gain) | 6-129481422 C > T | 0.01 (1/80) | 1/0 | 4.6 × 10−5 (7/152,130) | 0.004 | 0.003 (1/316) |
| rs121434505 | 8709 (1, P) | Fanconi anemia, complementation group E (AR) | FANCE (613976) | NM_021922.3: c.355C > T (NP_068741.1: p.Gln119Ter) | Nonsense (stop gain) | 6-35455853 C > T | 0.01 (1/80) | 1/0 | 6.6 × 10−5 (1/152,228) | 0.001 | 0.003 (1/316) |
| rs142637046 | 92361 (2, P) | Argininosuccinate lyase deficiency (AR) | ASL (608310) | NM_000048.4:c.446 + 1G > A | Splice donor | 7-66083175 G > A | 0.01 (1/80) | 1/0 | 7.9 × 10−5 (12/151,984) | 0.007 | 0.003 (1/316) |
| rs41286844 | 17038 (2, P) | Complement Component 6 deficiency (AR) | C8B (120960) | NM_000066.4:c.1282C > T (NP_000057.3:p.Arg428Ter) | Nonsense (stop gain) | 1-56940965 G > A | 0.03 (2/80) | 2/0 | 0.001 (195/151,786) | 0.005 | 0.02 (6/316) |
| rs1236367931 | 550946 (2, P) | Dysferlinopathy (AR) | DYSF (603009) | NM_007272.3:c.1471dup (NP_009203.2: p.Met491fs) | Frameshift | 2-71535283 G > GA | 0.08 (6/80) | 6/0 | 1.3 × 10−5 (2/152,020) | 4.9 × 10−19 | 0 (0/316) |
| rs7755898 | 12169 (2, P) | Classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency (AR) | CYP21A2 | NM_000500.9:c.955C > T (NP_000491.4:p.Gln319Ter) | Nonsense (stop gain) | 6-32040421 C > T | 0.08 (6/80) | 6/0 | 0.004 (523/136,986) | 7.6 × 10−7 | 0.004 (1/238) |
| rs202058123 | 430258 (1, LP) | Hereditary pancreatitis (AD) | CTRC (601405) | NM_007272.3:c.649G > A (NP_009203.2:p.Gly217Ser) | Missense | 1-15445606 G > A | 0.01 (1/80) | 1/0 | 6.6 × 10−5 (10/152,212) | 0.006 | 0.006 (2/316) |
| rs111033565 | 11876 (2, P) | Hereditary pancreatitis (AD) | PRSS1 (276000) | NM_002769.5:c.365G > A (NP_002760.1:p.Arg122His) | Missense | 7-142751938 G > A | 0.01 (1/80) | 1/0 | 5.5 × 10−5 (8/145,178) | 0.005 | 0 (0/302) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avnat, E.; Shapira, G.; Shoval, S.; Israel-Elgali, I.; Alkelai, A.; Shuldiner, A.R.; Gonzaga-Jauregui, C.; Zidan, J.; Maray, T.; Shomron, N.; et al. Comprehensive Genetic Analysis of Druze Provides Insights into Carrier Screening. Genes 2023, 14, 937. https://doi.org/10.3390/genes14040937
Avnat E, Shapira G, Shoval S, Israel-Elgali I, Alkelai A, Shuldiner AR, Gonzaga-Jauregui C, Zidan J, Maray T, Shomron N, et al. Comprehensive Genetic Analysis of Druze Provides Insights into Carrier Screening. Genes. 2023; 14(4):937. https://doi.org/10.3390/genes14040937
Chicago/Turabian StyleAvnat, Eden, Guy Shapira, Shelly Shoval, Ifat Israel-Elgali, Anna Alkelai, Alan R. Shuldiner, Claudia Gonzaga-Jauregui, Jamal Zidan, Taiseer Maray, Noam Shomron, and et al. 2023. "Comprehensive Genetic Analysis of Druze Provides Insights into Carrier Screening" Genes 14, no. 4: 937. https://doi.org/10.3390/genes14040937
APA StyleAvnat, E., Shapira, G., Shoval, S., Israel-Elgali, I., Alkelai, A., Shuldiner, A. R., Gonzaga-Jauregui, C., Zidan, J., Maray, T., Shomron, N., & Friedman, E. (2023). Comprehensive Genetic Analysis of Druze Provides Insights into Carrier Screening. Genes, 14(4), 937. https://doi.org/10.3390/genes14040937