
Netherton Syndrome Caused by Heterozygous Frameshift Mutation Combined with Homozygous c.1258A>G Polymorphism in SPINK5 Gene

 ,
,  ,
, 
 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
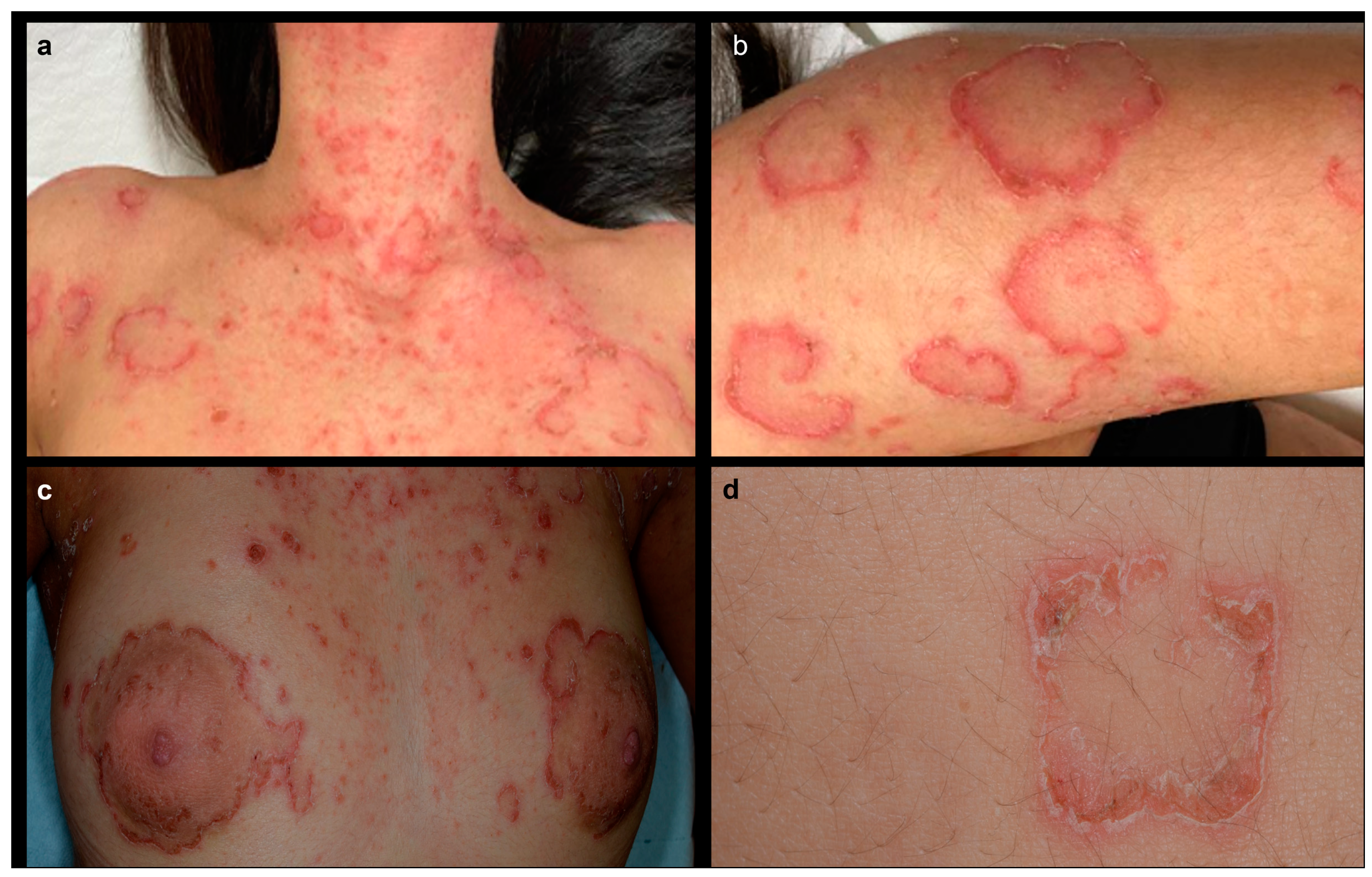
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, J.D.; Linden, K.G. Netherton syndrome: A case report and review of the literature. Int. J. Dermatol. 2006, 45, 693–697. [Google Scholar] [CrossRef]
- Sprecher, E.; Amin, S.; Nielsen, K.; Pfendner, E.; Uitto, J.; Richard, G.; Chavanas, S.; DiGiovanna, J.J.; Prendiville, J.S.; Silverman, R.; et al. The Spectrum of Pathogenic Mutations in SPINK5 in 19 Families with Netherton Syndrome: Implications for Mutation Detection and First Case of Prenatal Diagnosis. J. Investig. Dermatol. 2001, 117, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Bittencourt, M.D.J.S.; Moure, E.R.D.; Pies, O.T.C.; Mendes, A.D.; Deprá, M.M.; De Mello, A.L.P. Trichoscopy as a diagnostic tool in trichorrhexis invaginata and Netherton syndrome*. An. Bras. Dermatol. 2015, 90, 114–116. [Google Scholar] [CrossRef] [PubMed]
- SPINK5 Serine Peptidase Inhibitor Kazal Type 5 [Homo sapiens (human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/11005 (accessed on 10 May 2023).
- Sarri, C.A.; Roussaki-Schulze, A.; Vasilopoulos, Y.; Zafiriou, E.; Patsatsi, A.; Stamatis, C.; Gidarokosta, P.; Sotiriadis, D.; Sarafidou, T.; Mamuris, Z. Netherton Syndrome: A Genotype-Phenotype Review. Mol. Diagn. Ther. 2017, 21, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Chavanas, S.; Bodemer, C.; Rochat, A.; Hamel-Teillac, D.; Ali, M.; Irvine, A.; Bonafé, J.-L.; Wilkinson, J.; Taïeb, A.; Barrandon, Y.; et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat. Genet. 2000, 25, 141–142. [Google Scholar] [CrossRef]
- Stuvel, K.; Heeringa, J.J.; Dalm, V.A.S.H.; Meijers, R.W.J.; van Hoffen, E.; Gerritsen, S.A.M.; van Zelm, M.C.; Pasmans, S.G. Comel-Netherton syndrome: A local skin barrier defect in the absence of an underlying systemic immunodeficiency. Allergy 2020, 75, 1710–1720. [Google Scholar] [CrossRef]
- Walley, A.J.; Chavanas, S.; Moffatt, M.F.; Esnouf, R.M.; Ubhi, B.; Lawrence, R.; Wong, K.; Abecasis, G.R.; Jones, E.Y.; Harper, J.I.; et al. Gene polymorphism in Netherton and common atopic disease. Nat. Genet. 2001, 29, 175–178. [Google Scholar] [CrossRef]
- Nouwen, A.E.M.; Schappin, R.; Nguyen, N.T.; Ragamin, A.; Bygum, A.; Bodemer, C.; Dalm, V.A.S.H.; Pasmans, S.G.M.A. Outcomes of Systemic Treatment in Children and Adults With Netherton Syndrome: A Systematic Review. Front. Immunol. 2022, 13, 864449. [Google Scholar] [CrossRef]
- Barbati, F.; Giovannini, M.; Oranges, T.; Lodi, L.; Barni, S.; Novembre, E.; Baldo, E.; Cristofolini, M.; Stagi, S.; Ricci, S.; et al. Netherton Syndrome in Children: Management and Future Perspectives. Front. Pediatr. 2021, 9, 645259. [Google Scholar] [CrossRef]
- Leclerc-Mercier, S.; Bodemer, C.; Furio, L.; Hadj-Rabia, S.; de Peufeilhoux, L.; Weibel, L.; Bursztejn, A.-C.; Bourrat, E.; Ortonne, N.; Molina, T.J.; et al. Skin Biopsy in Netherton Syndrome: A Histological Review of a Large Series and New Findings. Am. J. Dermatopathol. 2016, 38, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.H.; Jacobson, K.A.; Rose, J.; Zeller, R. Hematoxylin and Eosin Staining of Tissue and Cell Sections. CSH Protoc. 2008, pdb.prot4986. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hamosh, A.; (Baltimore, MD, USA). Personal Communication, 2017.
- Kabesch, M.; Carr, D.; Weiland, S.K.; Von Mutius, E. Association between polymorphisms in serine protease inhibitor, kazal type 5 and asthma phenotypes in a large German population sample. Clin. Exp. Allergy 2004, 34, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Descargues, P.; Deraison, C.; Prost, C.; Fraitag, S.; Mazereeuw-Hautier, J.; D’Alessio, M.; Ishida-Yamamoto, A.; Bodemer, C. Corneodesmosomal Cadherins Are Preferential Targets of Stratum Corneum Trypsin- and Chymotrypsin-like Hyperactivity in Netherton Syndrome. J. Investig. Dermatol. 2006, 126, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Bitoun, E.; Micheloni, A.; Lamant, L.; Bonnart, C.; Tartaglia-Polcini, A.; Cobbold, C.; Al Saati, T.; Mariotti, F.; Mazereeuw-Hautier, J.; Boralevi, F.; et al. LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in Netherton syndrome. Hum. Mol. Genet. 2003, 12, 2417–2430. [Google Scholar] [CrossRef]
- Descargues, P.; Deraison, C.; Bonnart, C.; Kreft, M.; Kishibe, M.; Ishida-Yamamoto, A.; Elias, P.; Barrandon, Y.; Zambruno, G.; Sonnenberg, A.; et al. Spink5-deficient mice mimic Netherton syndrome through degradation of desmoglein 1 by epidermal protease hyperactivity. Nat. Genet. 2005, 37, 56–65. [Google Scholar] [CrossRef]
- Aoyama, Y.; Deraison, C.; Lacroix, M.; Bonnart, C.; Robin, A.; Besson, C.; Dubus, P.; Hovnanian, A. Faculty Opinions recommendation of Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J. Exp. Med. 2009, 206, 1135–1147. [Google Scholar] [CrossRef]
- Shimomura, Y.; Sato, N.; Kariya, N.; Takatsuka, S.; Ito, M. Netherton syndrome in two Japanese siblings with a novel mutation in the SPINK5 gene: Immunohistochemical studies of LEKTI and other epidermal molecules. Br. J. Dermatol. 2005, 153, 1026–1030. [Google Scholar] [CrossRef]
- Ong, C.; O’Toole, E.; Ghali, L.; Malone, M.; Smith, V.; Callard, R.; Harper, J. LEKTI demonstrable by immunohistochemistry of the skin: A potential diagnostic skin test for Netherton syndrome. Br. J. Dermatol. 2004, 151, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Hannula-Jouppi, K.; Laasanen, S.L.; Heikkilä, H.; Tuomiranta, M.; Tuomi, M.L.; Hilvo, S.; Kluger, N.; Kivirikko, S.; Hovnanian, A.; Mäkinen-Kiljunen, S.; et al. IgE allergen component-based profiling and atopic manifestations in patients with Netherton syndrome. J. Allergy Clin. Immunol. 2014, 134, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Schechter, N.M.; Brass, L.F.; Lavker, R.M.; Jensen, P.J. Reaction of mast cell proteases tryptase and chymase with protease activated receptors (PARs) on keratinocytes and fibroblasts. J. Cell. Physiol. 1998, 176, 365–373. [Google Scholar] [CrossRef]
- Thomas, W.R. Mite allergens groups I-VII. A catalogue of enzymes. Clin. Exp. Allergy 1993, 23, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qiu, F.; Wu, H.; Fan, Y.M. New compound heterozygous SPINK5 mutations in a Chinese infant with Netherton syndrome. J. Eur. Acad. Dermatol. Venereol. 2021, 35, e782–e784. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Lacaze-Buzy, L.; Furio, L.; Tron, E.; Valari, M.; Van der Wier, G.; Bodemer, C.; Bygum, A.; Bursztejn, A.-C.; Hovnanian, A.; et al. Clinical expression and new SPINK5 splicing defects in Netherton syndrome: Unmasking a frequent founder synonymous mutation and unconventional intronic mutations. J. Invest. Dermatol. 2012, 132, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Nevet, M.J.; Msc, M.I.; Ben-Ari, J.; Bergman, R. A case of Netherton syndrome with intestinal atresia, a novel SPINK5 mutation, and a fatal course. Int. J. Dermatol. 2017, 56, 1055–1057. [Google Scholar] [CrossRef]
- Guerra, L.; Fortugno, P.; Pedicelli, C.; Mazzanti, C.; Proto, V.; Zambruno, G.; Castiglia, D. Ichthyosis Linearis Circumflexa as the Only Clinical Manifestation of Netherton Syndrome. Acta Derm.-Venereol. 2015, 95, 720–724. [Google Scholar] [CrossRef]
- Di, W.-L.; Hennekam, R.; Callard, R.; Harper, J. A heterozygous null mutation combined with the G1258A polymorphism of SPINK5 causes impaired LEKTI function and abnormal expression of skin barrier proteins. Br. J. Dermatol. 2009, 161, 404–412. [Google Scholar] [CrossRef]
- Nishio, Y.; Noguchi, E.; Shibasaki, M.; Kamioka, M.; Ichikawa, E.; Ichikawa, K.; Umebayashi, Y.; Otsuka, F.; Arinami, T. Association between polymorphisms in the SPINK5 gene and atopic dermatitis in the Japanese. Genes Immun. 2003, 4, 515–517. [Google Scholar] [CrossRef]
- Kusunoki, T.; Okafuji, I.; Yoshioka, T.; Saito, M.; Nishikomori, R.; Heike, T.; Sugai, M.; Shimizu, A.; Nakahata, T. SPINK5 polymorphism is associated with disease severity and food allergy in children with atopic dermatitis. J. Allergy Clin. Immunol. 2005, 115, 636–638. [Google Scholar] [CrossRef]
- Fortugno, P.; Furio, L.; Teson, M.; Berretti, M.; El Hachem, M.; Zambruno, G.; Hovnanian, A.; D’Alessio, M. The 420K LEKTI variant alters LEKTI proteolytic activation and results in protease deregulation: Implications for atopic dermatitis. Hum. Mol. Genet. 2012, 21, 4187–4200. [Google Scholar] [CrossRef]
- Alpigiani, M.G.; Salvati, P.; Schiaffino, M.C.; Occella, C.; Castiglia, D.; Covaciu, C.; Lorini, R. A New SPINK5 Mutation in a Patient with Netherton Syndrome: A Case Report. Pediatr. Dermatol. 2012, 29, 521–522. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moltrasio, C.; Romagnuolo, M.; Riva, D.; Colavito, D.; Ferrucci, S.M.; Marzano, A.V.; Tadini, G.; Brena, M. Netherton Syndrome Caused by Heterozygous Frameshift Mutation Combined with Homozygous c.1258A>G Polymorphism in SPINK5 Gene. Genes 2023, 14, 1080. https://doi.org/10.3390/genes14051080
Moltrasio C, Romagnuolo M, Riva D, Colavito D, Ferrucci SM, Marzano AV, Tadini G, Brena M. Netherton Syndrome Caused by Heterozygous Frameshift Mutation Combined with Homozygous c.1258A>G Polymorphism in SPINK5 Gene. Genes. 2023; 14(5):1080. https://doi.org/10.3390/genes14051080
Chicago/Turabian StyleMoltrasio, Chiara, Maurizio Romagnuolo, Davide Riva, Davide Colavito, Silvia Mariel Ferrucci, Angelo Valerio Marzano, Gianluca Tadini, and Michela Brena. 2023. "Netherton Syndrome Caused by Heterozygous Frameshift Mutation Combined with Homozygous c.1258A>G Polymorphism in SPINK5 Gene" Genes 14, no. 5: 1080. https://doi.org/10.3390/genes14051080
APA StyleMoltrasio, C., Romagnuolo, M., Riva, D., Colavito, D., Ferrucci, S. M., Marzano, A. V., Tadini, G., & Brena, M. (2023). Netherton Syndrome Caused by Heterozygous Frameshift Mutation Combined with Homozygous c.1258A>G Polymorphism in SPINK5 Gene. Genes, 14(5), 1080. https://doi.org/10.3390/genes14051080