
Identification of LncRNAs and Functional Analysis of ceRNA Related to Fatty Acid Synthesis during Flax Seed Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
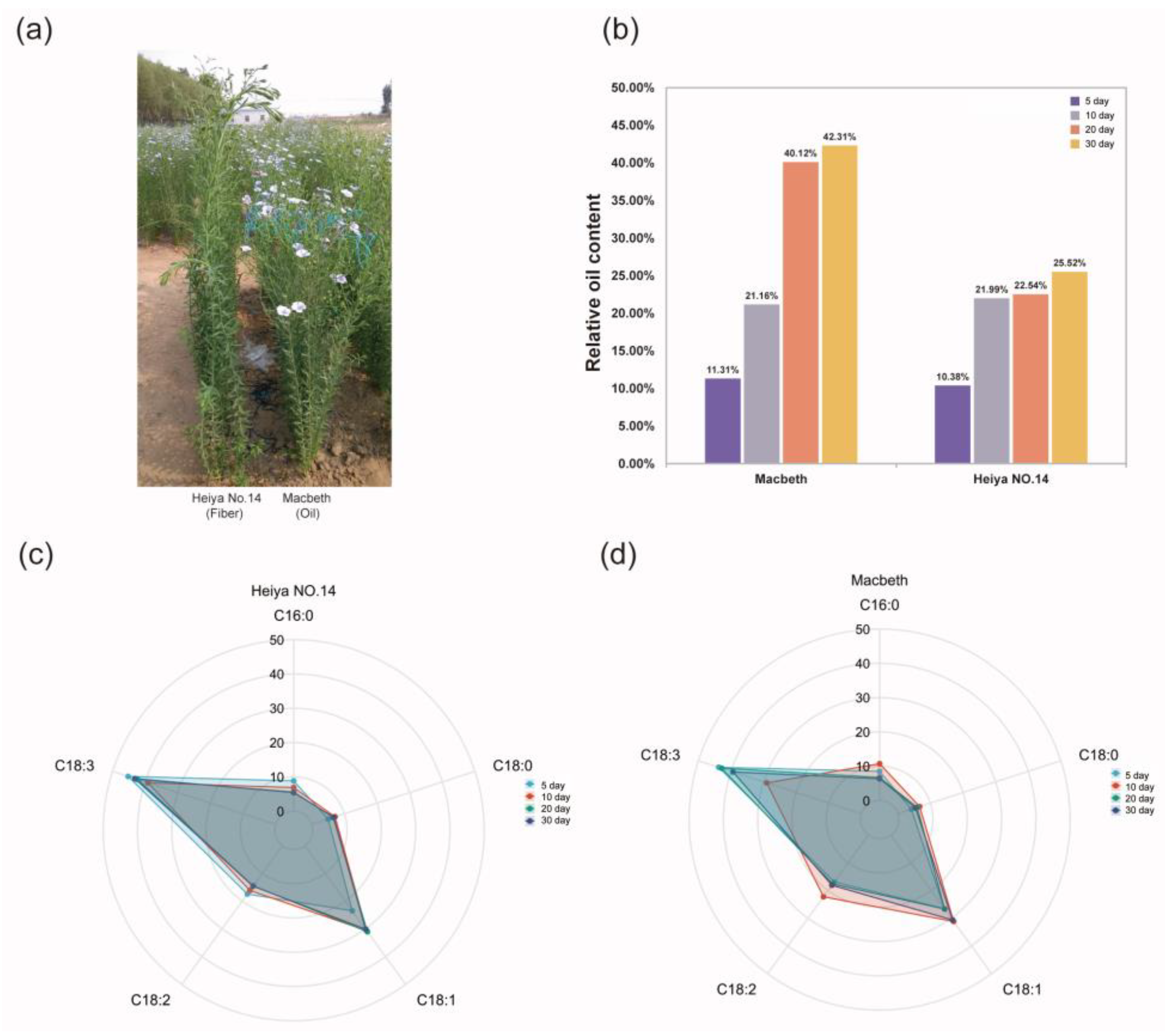
2.1. Sample Collection and Seed Oil Content Determination
2.2. RNA Extraction, Quality Control, and Sequencing
2.3. Strand-Specific Transcriptome Data Analysis
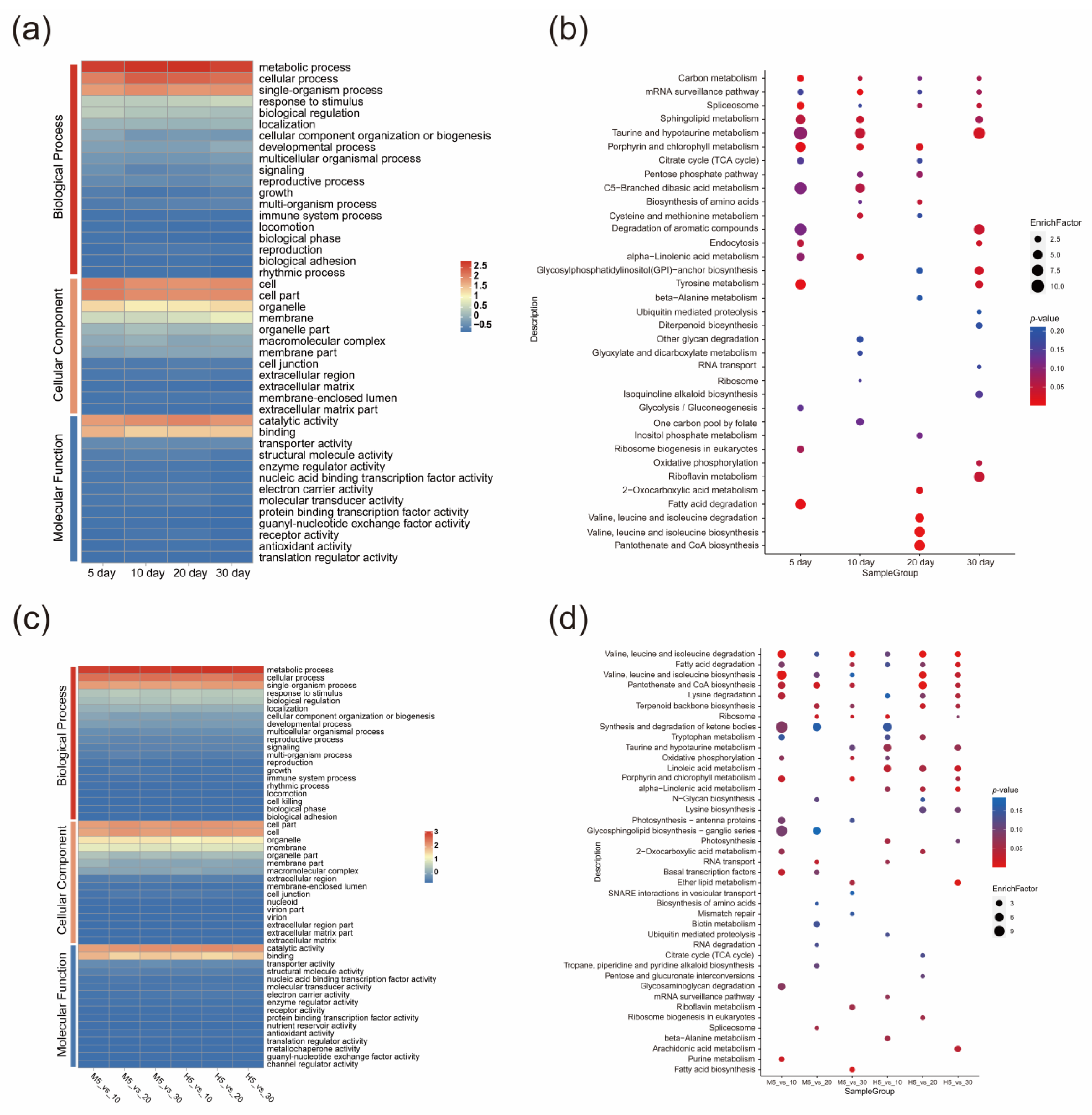
2.4. LncRNA Differential Expression Analysis and Functional Annotation
2.5. Construction of ceRNA Network Related to Seed Development
2.6. Validation of the ceRNAs Network by qRT-PCR
3. Results
3.1. Oil Content of Macbeth and Heiya NO.14 Varieties at Different Development Stages
3.2. Analysis of the Strand-Specific Transcriptome Data
3.3. Differential lncRNA Expression Analysis during Seed Development between the Macbeth and Heiya NO.14 Varieties
3.4. Construction and Validation of ceRNA Network Related to Seed Development in Macbeth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ludvíková, M.; Griga, M. Transgenic flax/linseed (Linum usitatissimum L.)—Expectations and reality. Czech J. Genet. Plant Breed. 2015, 51, 123–141. [Google Scholar] [CrossRef]
- Xie, D.; Dai, Z.; Yang, Z.; Tang, Q.; Deng, C.; Xu, Y.; Wang, J.; Chen, J.; Zhao, D.; Zhang, S.; et al. Combined genome-wide association analysis and transcriptome sequencing to identify candidate genes for flax seed fatty acid metabolism. Plant Sci. Int. J. Exp. Plant Biol. 2019, 286, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sha, L.; Li, Y.; Zhu, L.; Wang, Z.; Li, K.; Lu, H.; Bao, T.; Guo, L.; Zhang, X.; et al. Dietary α-Linolenic Acid-Rich Flaxseed Oil Exerts Beneficial Effects on Polycystic Ovary Syndrome Through Sex Steroid Hormones-Microbiota-Inflammation Axis in Rats. Front. Endocrinol. 2020, 11, 284. [Google Scholar] [CrossRef] [PubMed]
- Naghshi, S.; Aune, D.; Beyene, J.; Mobarak, S.; Asadi, M.; Sadeghi, O. Dietary intake and biomarkers of alpha linolenic acid and risk of all cause, cardiovascular, and cancer mortality: Systematic review and dose-response meta-analysis of cohort studies. BMJ Clin. Res. Ed. 2021, 375, n2213. [Google Scholar] [CrossRef]
- Alam, S.; Kim, M.; Shah, F.A.; Saeed, K.; Ullah, R.; Kim, M. Alpha-Linolenic Acid Impedes Cadmium-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration in Mouse Brain. Cells 2021, 10, 2274. [Google Scholar] [CrossRef]
- D’Angelo, S.; Motti, M.L.; Meccariello, R. ω-3 and ω-6 Polyunsaturated Fatty Acids, Obesity and Cancer. Nutrients 2020, 12, 2751. [Google Scholar] [CrossRef]
- Lu, X.; Chen, X.; Mu, M.; Wang, J.; Wang, X.; Wang, D.; Yin, Z.; Fan, W.; Wang, S.; Guo, L.; et al. Genome-Wide Analysis of Long Noncoding RNAs and Their Responses to Drought Stress in Cotton (Gossypium hirsutum L.). PLoS ONE 2016, 11, e156723. [Google Scholar] [CrossRef]
- Paralkar, V.R.; Taborda, C.C.; Huang, P.; Yao, Y.; Kossenkov, A.V.; Prasad, R.; Luan, J.; Davies, J.O.; Hughes, J.R.; Hardison, R.C.; et al. Unlinking an lncRNA from Its Associated cis Element. Mol. Cell 2016, 62, 104–110. [Google Scholar] [CrossRef]
- Zheng, X.M.; Chen, J.; Pang, H.B.; Liu, S.; Gao, Q.; Wang, J.R.; Qiao, W.H.; Wang, H.; Liu, J.; Olsen, K.M.; et al. Genome-wide analyses reveal the role of noncoding variation in complex traits during rice domestication. Sci. Adv. 2019, 5, x3619. [Google Scholar] [CrossRef] [PubMed]
- Latonen, L.; Afyounian, E.; Jylhä, A.; Nättinen, J.; Aapola, U.; Annala, M.; Kivinummi, K.K.; Tammela, T.T.L.; Beuerman, R.W.; Uusitalo, H.; et al. Integrative proteomics in prostate cancer uncovers robustness against genomic and transcriptomic aberrations during disease progression. Nat. Commun. 2018, 9, 1176. [Google Scholar] [CrossRef] [PubMed]
- Chua, B.A.; van der Werf, I.; Jamieson, C.; Signer, R.A.J. Post-Transcriptional Regulation of Homeostatic, Stressed, and Malignant Stem Cells. Cell Stem Cell 2020, 26, 138–159. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Zhang, A.; Zhao, Q.; Li, Z.; Lamboro, A.; He, H.; Li, Y.; Jiao, S.; Guan, S.; Liu, S.; et al. Genome-wide identification and analysis of long non-coding RNAs involved in fatty acid biosynthesis in young soybean pods. Sci. Rep. 2021, 11, 7603. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Liu, C.; Niu, X.; Wang, L.; Li, L.; Yuan, Q.; Pei, X. Research on lncRNA related to drought resistance of Shanlan upland rice. BMC Genom. 2022, 23, 336. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Li, S.; Hu, L.; Zhang, C. Genome-wide analysis of long non-coding RNAs (lncRNAs) in two contrasting rapeseed (Brassica napus L.) genotypes subjected to drought stress and re-watering. BMC Plant Biol. 2020, 20, 81. [Google Scholar] [CrossRef]
- Han, L.; Mu, Z.; Luo, Z.; Pan, Q.; Li, L. New lncRNA annotation reveals extensive functional divergence of the transcriptome in maize. J. Integr. Plant Biol. 2019, 61, 394–405. [Google Scholar] [CrossRef]
- Ren, J.; Jiang, C.; Zhang, H.; Shi, X.; Ai, X.; Li, R.; Dong, J.; Wang, J.; Zhao, X.; Yu, H. LncRNA-mediated ceRNA networks provide novel potential biomarkers for peanut drought tolerance. Physiol. Plant. 2022, 174, e13610. [Google Scholar] [CrossRef]
- De Carvalho, C.C.C.R.; Caramujo, M.J. The Various Roles of Fatty Acids. Molecules 2018, 23, 2583. [Google Scholar] [CrossRef] [PubMed]
- Heil, C.S.; Wehrheim, S.S.; Paithankar, K.S.; Grininger, M. Fatty Acid Biosynthesis: Chain-Length Regulation and Control. ChemBioChem Eur. J. Chem. Biol. 2019, 20, 2298–2321. [Google Scholar] [CrossRef]
- Russo, G.L. Dietary n-6 and n-3 polyunsaturated fatty acids: From biochemistry to clinical implications in cardiovascular prevention. Biochem. Pharmacol. 2009, 77, 937–946. [Google Scholar] [CrossRef]
- Tian, B.; Lu, T.; Xu, Y.; Wang, R.; Chen, G. Identification of genes associated with ricinoleic acid accumulation in Hiptage benghalensis via transcriptome analysis. Biotechnol. Biofuels 2019, 12, 16. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Gao, W.; Li, X.; Sun, S.; Xu, J.; Shi, X.; Guo, H. Regulatory mechanisms of fatty acids biosynthesis in Armeniaca sibirica seed kernel oil at different developmental stages. PeerJ 2022, 10, e14125. [Google Scholar] [CrossRef]
- Fofana, B.; Cloutier, S.; Duguid, S.; Ching, J.; Rampitsch, C. Gene expression of stearoyl-ACP desaturase and delta12 fatty acid desaturase 2 is modulated during seed development of flax (Linum usitatissimum). Lipids 2006, 41, 705–712. [Google Scholar] [CrossRef]
- Banik, M.; Duguid, S.; Cloutier, S. Transcript profiling and gene characterization of three fatty acid desaturase genes in high, moderate, and low linolenic acid genotypes of flax (Linum usitatissimum L.) and their role in linolenic acid accumulation. Genome 2011, 54, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Thambugala, D.; Duguid, S.; Loewen, E.; Rowland, G.; Booker, H.; You, F.M.; Cloutier, S. Genetic variation of six desaturase genes in flax and their impact on fatty acid composition. TAG. Theor. Angew. Genet. 2013, 126, 2627–2641. [Google Scholar] [CrossRef]
- Duguid, S.D.; Kenaschuk, E.O.; Rashid, K.Y. Macbeth flax. Can. J. Plant Sci. 2003, 83, 803–805. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Kang, Q.; Lu, Y.; Yang, X.; Guan, F.; Song, X. The Breeding Report of Heiya No. 14 of New Fiber Flax Variety. China Hemp Ind. 2003, 2003, 8–9. [Google Scholar]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2, accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.; Liu, X.; Zhao, S.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Guo, C.; Ma, X.; Xing, Y.; Zheng, C.; Xu, Y.; Shan, L.; Zhang, J.; Wang, S.; Wang, Y.; Carmichael, G.G.; et al. Distinct Processing of lncRNAs Contributes to Non-conserved Functions in Stem Cells. Cell 2020, 181, 621–636. [Google Scholar] [CrossRef]
- Deng, Y.; Li, J.; Wu, S.; Zhu, Y.; Chen, Y.; He, F. Integrated nr Database in Protein Annotation System and Its Localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Zhang, T.; Li, Z.; Song, X.; Han, L.; Wang, L.; Zhang, J.; Long, Y.; Pei, X. Identification and Characterization of microRNAs in the Developing Seed of Linseed Flax (Linum usitatissimum L.). Int. J. Mol. Sci. 2020, 21, 2708. [Google Scholar] [CrossRef]
- Li, Z.; Chi, H.; Liu, C.; Zhang, T.; Han, L.; Li, L.; Pei, X.; Long, Y. Genome-wide identification and functional characterization of LEA genes during seed development process in linseed flax (Linum usitatissimum L.). BMC Plant Biol. 2021, 21, 193. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Meng, X.; Li, A.; Yu, B.; Li, S. Interplay between miRNAs and lncRNAs: Mode of action and biological roles in plant development and stress adaptation. Comput. Struct. Biotec. 2021, 19, 2567–2574. [Google Scholar] [CrossRef] [PubMed]
- Green, A.G. Genetic control of polyunsaturated fatty acid biosynthesis in flax (Linum usitatissimum) seed oil. TAG. Theoretical and applied genetics. Theor. Angew. Genet. 1986, 72, 654–661. [Google Scholar] [CrossRef]
- Dang, Z.; Niu, J.; Dang, Z.; Sun, J.; Song, J.; Hao, R.; Li, W.; Wang, L. Accumulation of α-linolenic acid during seed development of Flax. Acta Agric. Boreali-Occident. Sin. 2014, 23, 90–95. [Google Scholar]
- Zhang, J.; Xu, H.; Yang, Y.; Zhang, X.; Huang, Z.; Zhang, D. Genome-wide analysis of long non-coding RNAs (lncRNAs) in two contrasting soybean genotypes subjected to phosphate starvation. BMC Genom. 2021, 22, 433. [Google Scholar] [CrossRef]
- Zhao, X.; Gan, L.; Yan, C.; Li, C.; Sun, Q.; Wang, J.; Yuan, C.; Zhang, H.; Shan, S.; Liu, J.N. Genome-Wide Identification and Characterization of Long Non-Coding RNAs in Peanut. Genes 2019, 10, 536. [Google Scholar] [CrossRef]
- Zheng, W.; Hu, H.; Lu, Q.; Jin, P.; Cai, L.; Hu, C.; Yang, J.; Dai, L.; Chen, J. Genome-Wide Identification and Characterization of Long Noncoding RNAs Involved in Chinese Wheat Mosaic Virus Infection of Nicotiana benthamiana. Biology 2021, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liao, J.; Li, Z.; Yu, Y.; Zhang, J.; Li, Q.; Qu, L.; Shu, W.; Chen, Y. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef]
- Benning, C. Mechanisms of lipid transport involved in organelle biogenesis in plant cells. Annu. Rev. Cell Dev. Biol. 2009, 25, 71–91. [Google Scholar] [CrossRef]
- Ohlrogge, J.; Browse, J. Lipid biosynthesis. Plant Cell 1995, 7, 957–970. [Google Scholar] [PubMed]
- Lee, G.; Zheng, Y.; Cho, S.; Jang, C.; England, C.; Dempsey, J.M.; Yu, Y.; Liu, X.; He, L.; Cavaliere, P.M.; et al. Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell 2017, 171, 1545–1558. [Google Scholar] [CrossRef]
- Ritte, G.; Lloyd, J.R.; Eckermann, N.; Rottmann, A.; Kossmann, J.; Steup, M. The starch-related R1 protein is an Alpha-glucan, water dikinase. Proc. Natl. Acad. Sci. USA 2002, 99, 7166–7171. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, R.; Baunsgaard, L.; Blennow, A. Functional characterization of Alpha-glucan, water dikinase, the starch phosphorylating enzyme. Biochem. J. 2004, 377 Pt 2, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Verma, A.K.; Kumar, V. Catalytic properties, functional attributes and industrial applications of β-glucosidases. 3 Biotech 2016, 6, 3. [Google Scholar] [CrossRef]
- Jiang, Z.; Long, L.; Liang, M.; Li, H.; Chen, Y.; Zheng, M.; Ni, H.; Li, Q.; Zhu, Y. Characterization of a glucose-stimulated β-glucosidase from Microbulbifer sp. ALW1. Microbiol. Res. 2021, 251, 126840. [Google Scholar] [CrossRef]
- Ischebeck, T.; Zbierzak, A.M.; Kanwischer, M.; Dörmann, P. A salvage pathway for phytol metabolism in Arabidopsis. J. Biol. Chem. 2006, 281, 2470–2477. [Google Scholar] [CrossRef]
- Compagnon, V.; Diehl, P.; Benveniste, I.; Meyer, D.; Schaller, H.; Schreiber, L.; Franke, R.; Pinot, F. CYP86B1 is required for very long chain omega-hydroxyacid and alpha, omega -dicarboxylic acid synthesis in root and seed suberin polyester. Plant Physiol. 2009, 150, 1831–1843. [Google Scholar] [CrossRef]
- Li, S.; Castillo-González, C.; Yu, B.; Zhang, X. The functions of plant small RNAs in development and in stress responses. Plant J. 2017, 90, 654–670. [Google Scholar] [CrossRef]
- Hou, H.; Li, J.; Gao, M.; Singer, S.D.; Wang, H.; Mao, L.; Fei, Z.; Wang, X. Genomic organization, phylogenetic comparison and differential expression of the SBP-box family genes in grape. PLoS ONE 2013, 8, e59358. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Cheng, J.; Ruan, S.; Qi, P.; Liu, W.; Bian, H.; Ye, L.; Zhang, Y.; Hu, J.; Dong, G.; et al. The heterochronic gene Oryza sativa LIKE HETEROCHROMATIN PROTEIN 1 modulates miR156b/c/i/e levels. J. Integr. Plant Biol. 2020, 62, 1839–1852. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H. The miR156/SPL Module, a Regulatory Hub and Versatile Toolbox, Gears up Crops for Enhanced Agronomic Traits. Mol. Plant. 2015, 8, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Feyissa, B.A.; Amyot, L.; Nasrollahi, V.; Papadopoulos, Y.; Kohalmi, S.E.; Hannoufa, A. Involvement of the miR156/SPL module in flooding response in Medicago sativa. Sci. Rep. 2021, 11, 3243. [Google Scholar] [CrossRef]
- Dong, H.; Yan, S.; Jing, Y.; Yang, R.; Zhang, Y.; Zhou, Y.; Zhu, Y.; Sun, J. MIR156-Targeted SPL9 Is Phosphorylated by SnRK2s and Interacts With ABI5 to Enhance ABA Responses in Arabidopsis. Front. Plant Sci. 2021, 12, 708573. [Google Scholar] [CrossRef]
- Wang, L.; Ming, L.; Liao, K.; Xia, C.; Sun, S.; Chang, Y.; Wang, H.; Fu, D.; Xu, C.; Wang, Z.; et al. Bract suppression regulated by the miR156/529-SPLs-NL1-PLA1 module is required for the transition from vegetative to reproductive branching in rice. Mol. Plant. 2021, 14, 1168–1184. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Bao, Q.; Mu, X.; Tong, C.; Cao, X.; Huang, J.; Xue, L.; Liu, C.; Fei, Y.; Loake, G.J. Glucose- and sucrose-signaling modules regulate the Arabidopsis juvenile-to-adult phase transition. Cell Rep. 2021, 36, 109348. [Google Scholar] [CrossRef]
- Rolland, F.; Winderickx, J.; Thevelein, J.M. Glucose-sensing mechanisms in eukaryotic cells. Trends Biochem. Sci. 2001, 26, 310–317. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Liu, C.; Tang, Q.; Zhang, T.; Wang, L.; Han, L.; Zhang, J.; Pei, X. Identification of LncRNAs and Functional Analysis of ceRNA Related to Fatty Acid Synthesis during Flax Seed Development. Genes 2023, 14, 967. https://doi.org/10.3390/genes14050967
Yang X, Liu C, Tang Q, Zhang T, Wang L, Han L, Zhang J, Pei X. Identification of LncRNAs and Functional Analysis of ceRNA Related to Fatty Acid Synthesis during Flax Seed Development. Genes. 2023; 14(5):967. https://doi.org/10.3390/genes14050967
Chicago/Turabian StyleYang, Xinsen, Caiyue Liu, Qiaoling Tang, Tianbao Zhang, Limin Wang, Lida Han, Jianping Zhang, and Xinwu Pei. 2023. "Identification of LncRNAs and Functional Analysis of ceRNA Related to Fatty Acid Synthesis during Flax Seed Development" Genes 14, no. 5: 967. https://doi.org/10.3390/genes14050967