
Peptides of a Feather: How Computation Is Taking Peptide Therapeutics under Its Wing
 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
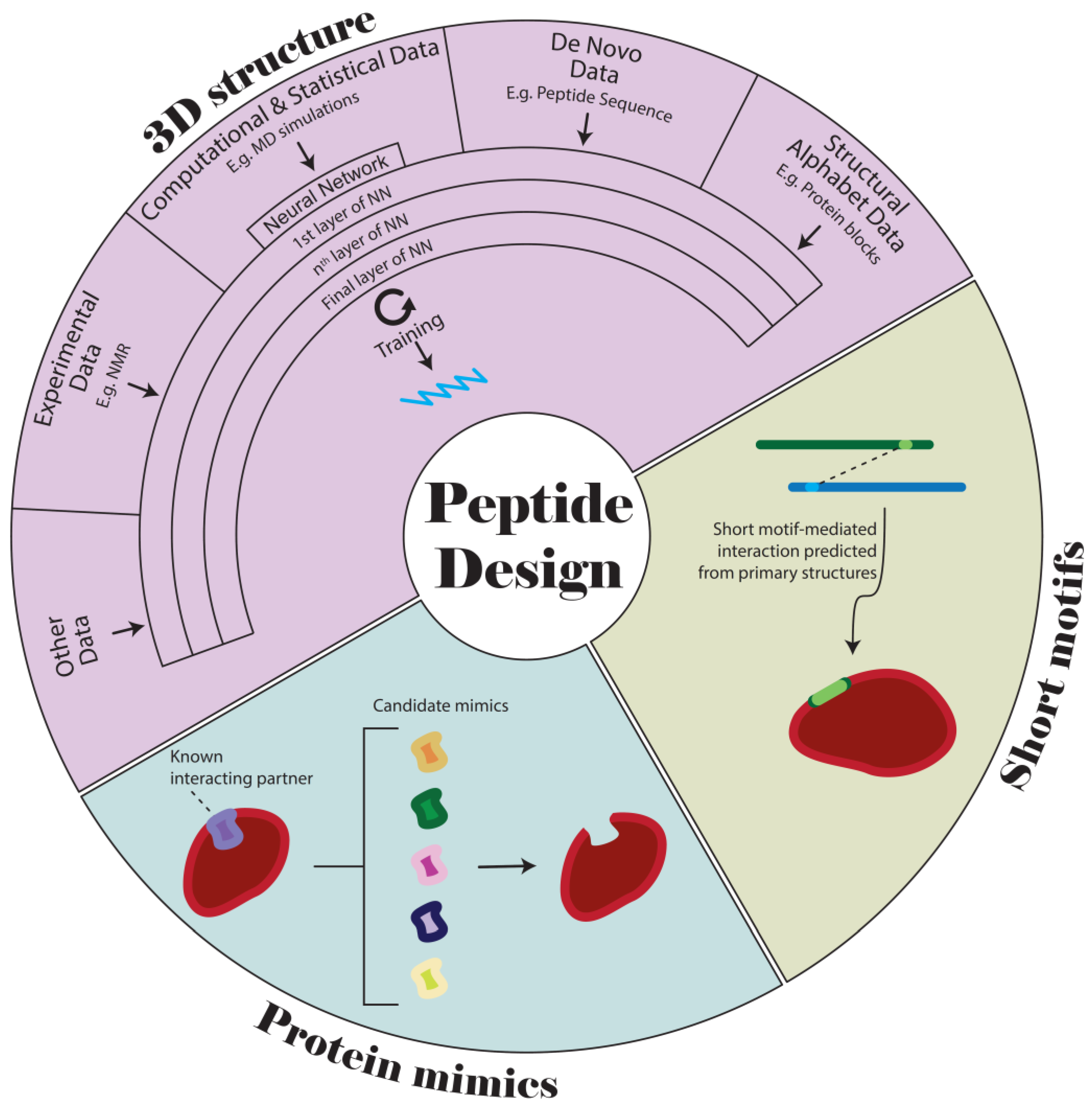
2. Algorithm and AI-Assisted Structural Design
2.1. Co-Evolutionary Analysis
2.2. Structural Alphabet
2.3. Deep Learning and Machine Learning
2.3.1. AlphaFold and AlphaDesign
2.3.2. Additional Neural Network-Harnessing Technologies
2.3.3. Rosetta
3. Designing Peptide Mimics
3.1. DeNovo
3.2. Structural-Guided Design
4. SM-Focused Design
4.1. Motif-Based Protein–Protein Interaction Prediction
4.2. SM-Based Peptide Design
5. Current Advantages and Future Challenges of In-Silico Peptide Design
5.1. Peptide Bioactivity and Toxicity
5.2. Barriers to FDA Approval of Computationally Designed Peptides
5.3. Applications of Therapeutic Peptides and the Future Peptide Design
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Basith, S.; Manavalan, B.; Shin, T.H.; Lee, G. Machine intelligence in peptide therapeutics: A next-generation tool for rapid disease screening. Med. Res. Rev. 2020, 40, 1276–1314. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic Peptides: Current Applications and Future Directions. Signal Transduct. Target Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Michael, S.; Alvarado-Valverde, J.; Mészáros, B.; Sámano-Sánchez, H.; Zeke, A.; Dobson, L.; Lazar, T.; Örd, M.; Nagpal, A.; et al. The eukaryotic linear motif resource: 2022 release. Nucleic Acids Res. 2022, 50, D497–D508. [Google Scholar] [CrossRef]
- Neduva, V.; Linding, R.; Su-Angrand, I.; Stark, A.; Masi, F.D.; Gibson, T.J.; Lewis, J.; Serrano, L.; Russell, R.B. Systematic discovery of new recognition peptides mediating protein interaction networks. PLoS Biol. 2005, 3, e405. [Google Scholar] [CrossRef]
- Jones, D.T.; Buchan, D.W.; Cozzetto, D.; Pontil, M. PSICOV: Precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics 2011, 28, 184–190. [Google Scholar] [CrossRef]
- Raybold, M.I.J.; Deane, C.M. The Therapeutic Antibody Profiler for Computational Developability Assessment. Ther. Antibodies Methods Protoc. 2022, 2313, 115–125. [Google Scholar]
- Schoenrock, A.; Samanfar, B.; Pitre, S.; Hooshyar, M.; Jin, K.; Phillips, C.A.; Wang, H.; Phanse, S.; Omidi, K.; Gui, Y.; et al. Efficient prediction of human protein-protein interactions at a global scale. BMC Bioinform. 2014, 15, 383. [Google Scholar] [CrossRef]
- Eid, F.-E.; ElHefnawi, M.; Heath, L.S. DeNovo: Virus-host sequence-based protein-protein interaction predition. Bioinformatics 2016, 32, 1144–1150. [Google Scholar] [CrossRef]
- Burnside, D.; Schoenrock, A.; Moteshareie, H.; Hooshyar, M.; Basra, P.; Hajikarimlou, M.; Dick, K.; Barnes, B.; Kazmirchuk, T.; Jessulat, M.; et al. In Silico Engineering of Synthetic Binding Proteins from Random Amino Acid Sequences. iScience 2018, 11, 375–387. [Google Scholar] [CrossRef]
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Huang, G.; Liao, W.; Gong, S.; Xiao, J.; Bai, J.; Hsiao, W.L.W.; Li, N.; Wu, J.-L. Discovery of the bioactive peptides secreted by Bifidobacterium using integrated MCX coupled with LC-MS and feature-based Molecular Networking. Food Chem. 2021, 347, 129008. [Google Scholar] [CrossRef]
- Eppelmann, K.; Doekel, S.; Marahiel, M.A. Engineered biosynthesis of the peptide antibiotic bacitracin in the surrogate host bacillus subtilis. J. Biol. Chem. 2001, 276, 34823–34831. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Azadvari, N.; Hosseinzadeh, P. Design of Protein Segments and Peptides for Binding to Protein Targets. BioDesign Res. 2022, 2022, 9783197. [Google Scholar] [CrossRef]
- Kim, J.; Lim, T.Y.; Park, J.; Jang, Y. Recombinant Protein Mimicking the Antigenic Structure of the Viral Surface Envelope Protein Reinforces the Induction of an Antigen-Specific and Virus-Neutralizing Immune Response against the Dengue Virus. J. Microbiol. 2022, 61, 131–143. [Google Scholar] [CrossRef]
- Kobayakawa, T.; Yokoyama, M.; Tsuji, K.; Fujino, M.; Kurakami, M.; Onishi, T.; Boku, S.; Ishii, T.; Miura, Y.; Shinohara, K.; et al. Low-Molecular-Weight Anti-HIV-1 Agents Targeting HIV-1 Capsid Proteins. RSC Adv. 2023, 13, 2156–2167. [Google Scholar] [CrossRef]
- Qui, Y.; Wei, G. Persistent Spectral Theory-Guided Protein Engineering. Nat. Comput. Sci. 2023, 3, 149–163. [Google Scholar]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef]
- Ceol, A.; Chatr-aryamontri, A.; Santonico, E.; Sacco, R.; Castagnoli, L.; Cesareni, G. DOMINO: A database of domain–peptide interactions. Nucleic Acids Res. 2007, 35 (Suppl. S1), D557–D560. [Google Scholar] [CrossRef]
- Pawson, T.; Nash, P. Assembly of cell regulatory systems through protein interaction domains. Science 2003, 300, 445–452. [Google Scholar] [CrossRef]
- Shell, M.S.; Ozkan, S.B.; Voelz, V.A.; Wu, G.A.; Dill, K.A. Blind Test of Physics-Based Prediction of Protein Structures. Biophys. J. 2009, 96, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Karplus, P.A. A fresh look at the Ramachandran plot and the occurrence of standard structures in proteins. Biomol. Concepts 2010, 1, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.P.; Singh, K.; Hazra, A.; Madhusudhan, M.S. Peptide bond planarity constrains hydrogen bond geometry and influences secondary structure conformations. Curr. Res. Struct. Biol. 2021, 3, 1–8. [Google Scholar] [CrossRef]
- Jendrusch, M.; Korbel, J.O.; Sadiq, S.K. AlphaDesign: A de novo protein design framework based on AlphaFold. bioRxiv 2021, 1–20. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, C.; Liu, H. Fragment-based local statistical potentials derived by combining an alphabet of protein local structures with secondary structures and solvent accessibilities. Proteins 2009, 74, 820–836. [Google Scholar] [CrossRef] [PubMed]
- Ołdziej, S.; Czaplewski, C.; Liwo, A.; Chinchio, M.; Nanias, M.; Vila, J.; Khalili, M.; Arnautova, Y.A.; Jagielska, A.; Makowski, M.A.; et al. Physics-based protein-structure prediction using a hierarchical protocol based on the UNRES force field: Assessment in two blind tests. Proc. Natl. Acad. Sci. USA 2005, 102, 7547–7552. [Google Scholar] [CrossRef]
- Knapp, B.; Demharter, S.; Esmaielbeiki, R.; Deane, C.M. Current status and future challenges in T-cell receptor/peptide/MHC molecular dynamics simulations. Brief Bioinform. 2015, 16, 1035–1044. [Google Scholar] [CrossRef]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Childers, M.C.; Daggett, V. Validating molecular dynamics simulations against experimental observables in light of underlying conformational ensembles. J. Phys. Chem. 2018, 122, 6673–6689. [Google Scholar] [CrossRef]
- Pearce, R.; Huang, X.; Omenn, G.S.; Zhang, Y. De novo protein fold design through sequence-independent fragment assembly simulations. Proc. Natl. Acad. Sci. USA 2023, 120, e2208275120. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Kuhlman, B.; Dantas, G.; Ireton, G.C.; Varani, G.; Stoddard, B.L.; Baker, D. Design of a novel globular protein fold with atomic-level accuracy. Science 2003, 302, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, A.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Thorpe, I.F.; Izvekov, S.; Voth, G.A. Coarse-grained peptide modeling using a systematic multiscale approach. Biophys. J. 2007, 92, 4289–4303. [Google Scholar] [CrossRef] [PubMed]
- Shmilovich, K.; Mansbach, R.A.; Sidky, H.; Dunne, O.E.; Panda, S.S.; Tovar, J.D.; Ferguson, A.L. Discovery of Self-assembling pi-conjugated peptides by active learning-directed coarse-grained molecular simulation. J. Phys. Chem. 2020, 124, 3873–3891. [Google Scholar] [CrossRef]
- Oteri, F.; Nadalin, F.; Champeimont, R.; Carbone, A. BIS2Analyzer: A server for co-evolution analysis of conserved protein families. Nucleic Acids Res. 2017, 45, 307–314. [Google Scholar] [CrossRef]
- Petrov, P.B.; Awoniyi, L.O.; Šuštar, V.; Balci, M.Ö.; Mattila, P.K. AutoCoEv—A high-throughput in silico pipeline for predicting inter-protein coevolution. Int. J. Mol. Sci. 2022, 23, 3351. [Google Scholar] [CrossRef]
- Etchebest, C.; Benros, C.; Hazout, S.; de Brevern, A.G. A structural alphabet for local protein structures: Improved prediction methods. Proteins Struct. Funct. Genet. 2005, 59, 810–827. [Google Scholar] [CrossRef]
- Craveur, P.; Joseph, A.P.; Esque, J.; Narwani, T.J.; Noel, F.; Shinada, N.K.; Goguet, M.; Leonard, S.; Poulain, P.; Bertrand, O.F.; et al. Protein flexibility in the light of structural alphabets. Front. Mol. Biosci. 2015, 2, 20. [Google Scholar] [CrossRef]
- Shapovalov, M.V.; Dunbrack, R.L. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. Pep-fold3: Faster de novostructure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.-F.; Zhou, X.-X.; Willems, S.; Ammar, C.; Wahle, M.; Bludau, I.; Voytik, E.; Strauss, M.T.; Mann, M. Alphapeptdeep: A modular deep learning framework to predict peptide properties for proteomics. Nat. Commun. 2022, 13, 7238. [Google Scholar] [CrossRef] [PubMed]
- Upmeier zu Belzen, J.; Bürgel, T.; Holderbach, S.; Bubeck, F.; Adam, L.; Gandor, C.; Klein, M.; Mathony, J.; Pfuderer, P.; Platz, L.; et al. Leveraging implicit knowledge in neural networks for functional dissection and engineering of proteins. Nat. Mach. Intell. 2019, 1, 225–235. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Wang, C.; Lo, C.C.; Liu, X.; Wu, W.; Zhang, J. ProDCoNN: Protein design using a convolutional neural network. Proteins Struct. Funct. Genet. 2020, 88, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Norn, C.; Wicky, B.I.; Juergens, D.; Liu, S.; Kim, D.; Tischer, D.; Koepnick, B.; Anishchenko, I.; Baker, D. Protein sequence design by conformational landscape optimization. Proc. Natl. Acad. Sci. USA 2021, 118, e2017228118. [Google Scholar] [CrossRef]
- Wang, S.; Peng, J.; Ma, J.; Xu, J. Protein secondary structure prediction using deep convolutional Neural Fields. Sci. Rep. 2016, 6, 18962. [Google Scholar] [CrossRef]
- Zhang, H.; Huang, Y.; Bei, Z.; Ju, Z.; Meng, J.; Hao, M.; Zhang, J.; Zhang, H.; Xi, W. Inter-residue distance prediction from Duet Deep Learning Models. Front. Genet. 2022, 13, 887491. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with alphafold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- McDonald, E.F.; Jones, T.; Plate, L.; Meiler, J.; Gulsevin, A. Benchmarking alphafold2 on peptide structure prediction. Structure 2023, 31, 111–119. [Google Scholar] [CrossRef]
- Tsaban, T.; Varga, J.K.; Avraham, O.; Ben-Aharon, Z.; Khramushin, A.; Schueler-Furman, O. Harnessing protein folding neural networks for peptide–protein docking. Nat Commun. 2022, 13, 176. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, X.; Chen, Q.; Pan, B.; Lou, J.; Jia, Z.; Du, Y.; Xu, W.; Zhang, L.; Feng, X.; et al. Novel Muscle-Homing Peptide FGF1 Conjugate Based on AlphaFold for Type 2 Diabetes Mellitus. ACS Appl. Mater. Interfaces 2023, 15, 6397–6410. [Google Scholar] [CrossRef] [PubMed]
- Modi, R.; McKee, N.; Zhang, N.; Alwali, A.; Nelson, A.; Lohar, A.; Ostafe, R.; Zhang, D.D.; Parkinson, E.I. Stapled Peptides as Direct Inhibitors of Nrf2-sMAF Transcription Factors. J. Med. Chem. 2023, 66, 6184–6192. [Google Scholar] [CrossRef] [PubMed]
- Bertoline, L.M.F.; Lima, A.N.; Krieger, J.E.; Teixeira, S.K. Before and after AlphaFold2: An overview of protein structure prediction. Front. Bioinform. 2023, 3, 1120370. [Google Scholar] [CrossRef]
- Wolfe, M.; Webb, S.; Chushak, Y.; Krabacher, R.; Liu, Y.; Swami, N.; Harbaugh, S.; Chavez, J. A high-throughput pipeline for design and selection of peptides targeting the SARS-CoV-2 Spike protein. Sci. Rep. 2021, 11, 21768. [Google Scholar] [CrossRef]
- Gunderwala, A.Y.; Nimbvikar, A.A.; Cope, N.J.; Li, Z.; Wang, Z. Development of Allosteric BRAF Peptide Inhibitors Targeting the Dimer Interface of BRAF. ACS Chem. Biol. 2019, 14, 1471–1480. [Google Scholar] [CrossRef]
- Xing, R.; Guo, Z.; Lu, H.; Zhang, Q.; Liu, Z. Molecular Imprinting and Cladding Produces Antibody Mimics with Significantly Improved Affinity and Specificity. Sci. Bull. 2022, 67, 278–287. [Google Scholar] [CrossRef]
- Wang, X.; Guillem, J.M.; Lee, D.; Hinck, A.; Beth, R.; Baker, D. De Novo Design of BMP Mimics. Biophys. J. 2023, 122, 312a. [Google Scholar] [CrossRef]
- Park, J.S.; Choi, J.; Cao, L.; Mohanty, J.; Suzuki, Y.; Park, A.; Baker, D.; Schlessinger, J.; Lee, S. Isoform-Specific Inhibition of FGFR Signalling Achieved by b De-Novo-Designed Mini Protein. Cell Rep. 2022, 41, 111545. [Google Scholar] [CrossRef]
- Levy, S.; Somasundaram, L.; Raj, I.X.; Ic-Mex, D.; Phal, A.; Schmidt, S.; Ng, W.I.; Mar, D.; Decarreau, J.; Moss, N.; et al. Dcas9 Fusion to Computer-Designed PRC2 Inhibitor Reveals Functional TATA Box in Distal Promotor Region. Cell Rep. 2022, 38, 110457. [Google Scholar] [CrossRef]
- Kaur, G.; Kapoor, S.; Kaundal, S.; Dutta, D.; Thakur, K.G. Structure-Guided Designing and Evaluation of Peptides Targeting Bacterial Transcription. Front. Bioeng. Biotechnol. 2020, 8, 797. [Google Scholar] [CrossRef] [PubMed]
- Karoyan, P.; Vieillard, V.; Gómez-Morales, L.; Odile, E.; Guihot, A.; Luyt, C.-E.; Denis, A.; Grondin, P.; Lequin, O. Human ACE2 peptide-mimics block SARS-CoV-2 pulmonary cells infection. Commun. Biol. 2021, 4, 197. [Google Scholar] [CrossRef] [PubMed]
- Cardote, T.A.F.; Ciulli, A. Structure-Guided Design of Peptides as Tools to Probe the Protein–Protein Interaction between Cullin-2 and Elongin BC Substrate Adaptor in Cullin RING E3 Ubiquitin Ligases. Chem. Med. Chem. 2017, 12, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Hellman, L.M.; Foley, K.C.; Singh, N.K.; Alonso, J.A.; Riley, T.P.; Devlin, J.R.; Ayres, C.M.; Keller, G.L.J.; Zhang, Y.; Kooi, C.W.V.; et al. Improving T Cell Receptor On-Target Specificity via Structure-Guided Design. Mol. Ther. 2023, 27, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Merritt, H.I.; Sawyer, N.; Watkins, A.M.; Arora, P.S. Anchor residues Govern Binding and Folding of An Intrinsically Disordered Domain. ACS Chem. Biol. 2022, 17, 2723–2727. [Google Scholar] [CrossRef]
- Bansata, B.; Bick, M.J.; Bera, A.K.; Norn, C.; Chow, C.M.; Carter, L.P.; Goreshnik, I.; Diamaio, F.; Baker, D. An enumerative algorithm for de novo design of proteins with diverse pocket structures. Proc. Natl. Acad. Sci. USA 2020, 117, 22135–22145. [Google Scholar]
- Hesslow, D.; Zanichelli; Notin, P.; Poli, I.; Marks, D. RITA: A Study on Scaling Up Generative Protein Sequence Models. arXiv 2022, arXiv:2205.05789. [Google Scholar]
- Pitre, S.; Dehne, F.; Chan, A.; Cheetham, J.; Duong, A.; Emili, A.; Gebbia, M.; Greenblatt, J.; Jessulat, M.; Krogan, N.; et al. PIPE: A protein-protein interaction prediction engine based on the re-occurring short polypeptide sequences between known interacting protein pairs. BMC Bioinform. 2006, 7, 365. [Google Scholar] [CrossRef]
- Pitre, S.; Alamgir, M.; Green, J.R.; Dumontier, M.; Dehne, F.; Golshani, A. Computational methods for predicting protein-protein interactions. Adv. Biochem. Eng. Biotechnol. 2008, 110, 247–267. [Google Scholar]
- Amos-Binks, A.; Patulea, C.; Pitre, S.; Schoenrock, A.; Gui, Y.; Green, J.R.; Golshani, A.; Dehne, F. Binding site prediction for protein-protein interactions and novel motif discovery using re-occurring polypeptide sequences. BMC Bioinform. 2011, 12, 225. [Google Scholar] [CrossRef]
- Pitre, S.; Hooshyar, M.; Schoenrock, A.; Samanfar, B.; Jessulat, M.; Green, J.R.; Dehne, F.; Golshani, A. Short Co-occurring Polypeptide Regions Can Prediction Global Protein Interaction Maps. Sci. Rep. 2012, 2, 239. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Heo, L.; Lee, M.S.; Seok, C. GalaxyPepDock: A protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 2015, 6, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Neduva, V.; Russell, R.B. DILMOT: Discovery of linear motifs in proteins. Nucleic Acids Res. 2006, 34, W350–W355. [Google Scholar] [CrossRef] [PubMed]
- Krystowiak, I.; Davey, N.E. SLiMSearch: A framework for proteome-wide discovery and annotation of functional modules in intrinsically disordered regions. Nucleic Acids Res. 2017, 45, W464–W469. [Google Scholar] [CrossRef]
- Ghoorah, A.W.; Devignes, M.-D.; Smail-Tabbone, M.; Ritchie, D.W. KBDOCK 2013: A spatial classification of 3D protein domain family interactions. Nucleic Acids Res. 2014, 42, D389–D395. [Google Scholar] [CrossRef]
- Stein, A.; Russell, R.B.; Aloy, P. 3did: Interacting protein domains of known three-dimensional structure. Nucleic Acids Res. 2005, 33, D413–D417. [Google Scholar] [CrossRef]
- Ben-Hur, A.; Noble, W.S. Choosing negative examples for the prediction of protein-protein interactions. BMC Bioinform. 2006, 7, S2. [Google Scholar] [CrossRef]
- Dick, K.; Samanfar, B.; Barnes, B.; Cober, E.R.; Mimee, B.; Tan, L.H.; Molnar, S.J.; Biggar, K.K.; Golshani, A.; Dehne, F.; et al. PIPE4: Fast PPI predictor for comprehensive inter- and cross-species interactions. Sci. Rep. 2020, 10, 1390. [Google Scholar] [CrossRef]
- Barnes, B.; Karimloo, M.; Schoenrock, A.; Burnside, D.; Cassol, E.; Wong, A.; Dehne, F.; Golshani, A.; Green, J.R. Predicting novel protein-protein interactions between the HIV-1 virus and homo sapiens. In Proceedings of the IEEE EMBS, Orlando, FL, USA, 16–20 August 2016; pp. 1–4. [Google Scholar]
- Samanfar, B.; Shostak, K.; Moteshareie, H.; Hajikarimlou, M.; Shaikho, S.; Omidi, K.; Hooshyar, M.; Burnside, D.; Galvan Marques, I.; Kazmirchuk, T.; et al. The sensitivity of the yeast, Saccharomyces cerevisiae, to acetic acid is influenced by DOM34 and RPL36A. PeerJ 2017, 5, e4037. [Google Scholar] [CrossRef]
- Kazmirchuk, T.; Dick, K.; Burnside, D.J.; Barnes, B.; Moteshareie, H.; Hajikarimlou, M.; Omidi, K.; Ahmed, D.; Low, A.; Lettl, C.; et al. Designing anti-Zika virus peptides derived from predicted human-Zika virus protein-protein interactions. Comput. Biol. Chem. 2017, 71, 180–187. [Google Scholar] [CrossRef]
- Dick, K.; Green, J.R. Reciprocal perspective for improved protein-protein interaction prediction. Sci. Rep. 2018, 8, 11964. [Google Scholar] [CrossRef] [PubMed]
- Dick, K.; Pattang, A.; Hooker, J.; Nissan, N.; Sadowski, M.; Barnes, B.; Tan, L.H.; Burnside, D.; Phanse, S.; Aoki, H.; et al. Human-Soybean Allergies: Elucidation of Seed Proteome and Comprehensive Protein-Protein Interaction Prediction. J. Proteome Res. 2021, 20, 4925–4947. [Google Scholar] [CrossRef]
- Hajikarimlou, M.; Hooshyar, M.; Moutaoufik, M.T.; Aly, K.A.; Azad, T.; Takallou, S.; Jagadeesan, S.; Phanse, S.; Said, K.B.; Samanfar, B.; et al. A computational approach to rapidly design peptides that detect SARS-CoV-2 surface protein S. NAR Genom. Bioinform. 2022, 4, lqac058. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Banting, F.G.; Best, H.C.; Collip, J.B.; Macleod, J.J.R.; Noble, E.C. The effect of pancreatic extract (insulin) on normal rabbits. Am. J. Physiol. 1922, 62, 162–176. [Google Scholar] [CrossRef]
- Win, T.S.; Schaduangrat, N.; Prachayasittikul, V.; Nantasenamat, C.; Shoombuatong, W. PAAP: A web server for predicting antihypertensive activity of peptides. Future Med. Chem. 2018, 10, 1749–1767. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Lin, Y.; Ding, Y.; Wang, Y.; Cheng, X.; Lin, Z. QSAR study on angiotensin-converting enzyme inhibitor oligopeptides based on a novel set of sequence information descriptors. J. Mol. Model. 2011, 17, 1599–1606. [Google Scholar] [CrossRef]
- Kumar, R.; Chaudhary, K.; Singh Chauhan, J.; Nagpal, G.; Kumar, R.; Sharma, M.; Raghava, G.P. An in silico platform for predicting, screening, and designing antihypertensive peptides. Sci. Rep. 2015, 5, 12512. [Google Scholar] [CrossRef]
- Win, T.S.; Malik, A.A.; Prachayasittikul, V.; Wikberg, J.E.S.; Nantasenamat, C.; Shoombuatong, W. HemoPred: A web server for predicting the hemolytic activity of peptides. Future Med. Chem. 2017, 9, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Jing, R.; Liu, F.; Luo, J.; Li, Y. DeepACP: A Novel Computational Approach for Accurate Identification of Anticancer Peptides by Deep Learning Algorithm. Mol. Ther. Nucleic Acids 2020, 22, 862–870. [Google Scholar] [CrossRef]
- Boyanapalli, R.; Singh, I.; Faria, M. Peptides and Oligonucleotide-Based Therapy: Bioanalytical Challenges and Practical Solutions. In An Introduction to Bioanalysis of Biopharmaceuticals; Kumar, S., Ed.; AAPS Advances in the Pharmaceutical Sciences Series; Springer: Cham, Switzerland, 2022; Volume 57, pp. 131–155. [Google Scholar]
- Castillo, F.; Corbi-Verge, C.; Murciano-Calles, J.; Candel, A.M.; Han, Z.; Iglesias-Bexiga, M.; Ruiz-Sanz, J.; Kim, P.M.; Harty, R.N.; Martinez, J.C.; et al. Phage Display Identification of Nanomolar Ligands for Human NEDD4-WW3: Energetic and Dynamic Implications for the Development of Broad-Spectrum Antivirals. Int. J. Biol. Macromol. 2022, 207, 308–323. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, V.; Jain, P.; Akhtar, N.; Joshi, A.; Gupta, L.R.; Grewal, R.K.; Oliva, R.; Shaikh, A.R.; Cavallo, L.; Chawla, M. Immunoinformatics-Aided Design and In Vivo Validation of a Peptide-Based Multiepitope Vaccine Targeting Canine Circovirus. ACS Pharmacol. Transl. Sci. 2022, 5, 679–691. [Google Scholar] [CrossRef]
- Lohia, N.; Baranwal, M. An Immunoinformatics approach in the design of synthetic peptide vaccine against influenza virus. Immunoinformatics 2020, 2131, 229–243. [Google Scholar]
- Dong, H.L.; Sui, Y.F. Prediction of HLA-A2-restricted CTL epitope specific to HCC by SYFPEITHI combined with polynomial method. World J. Gastroenterol. 2005, 11, 208–211. [Google Scholar] [CrossRef]
- Nielsen, M.; Andreatta, M.; Peters, B.; Buus, S. Immunoinformatics: Predicting peptide-MHC binding. Annu. Rev. Biomed. Data Sci. 2020, 3, 191–215. [Google Scholar] [CrossRef]
- Peters, B.; Tong, W.; Sidney, J.; Sette, A.; Weng, Z. Examining the independent binding assumption for binding of peptide epitopes to MHC-I molecules. Bioinformatics 2003, 19, 1765–1772. [Google Scholar] [CrossRef]
- Peters, B.; Sette, A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinform. 2005, 6, 132. [Google Scholar] [CrossRef]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef]
- Yao, Y.; Du, X.; Diao, Y.; Zhu, H. An integration of deep learning with feature embedding for protein-protein interaction prediction. PeerJ 2019, 7, e7126. [Google Scholar] [CrossRef]
- Charih, F.; Biggar, K.K.; Green, J.R. Assessing sequence-based protein–protein interaction predictors for use in therapeutic peptide engineering. Sci. Rep. 2022, 12, 9610. [Google Scholar] [CrossRef]
- Ramelot, T.A.; Palmer, J.; Montelione, G.T.; Bhardwaj, G. Cell-Permeable Chameleonic Peptides: Exploiting Conformational Dynamics in De Novo Cyclic Peptide Design. Curr. Opin. Struct. Biol. 2023, 80, 102603. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Peng, X.; Leal, J.; Mohanty, R.P. Peptides as drug delivery vehicles across biological barriers. J. Pharm. Investig. 2018, 48, 89–111. [Google Scholar] [CrossRef] [PubMed]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Basics and Recent Advances in Peptide and Protein Drug Delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef] [PubMed]
- Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef]
- Cooper, B.M.; Iegre, J.; O’Donovan, D.H.; Halvarsson, M.Ö.; Spring, D.R. Peptides as a Platform for Targeted Therapeutics for Cancer: Peptide-Drug Conjugates (PDCs). Chem. Soc. Rev. 2021, 50, 1–15. [Google Scholar] [CrossRef]
- Verma, S.; Goand, U.M.; Husain, A.; Katekar, R.A.; Garg, R.; Gayen, J.R. Challenges of peptide and protein drug delivery by oral route: Current strategies to improve bioavailability. Drug Dev. Res. 2021, 82, 927–944. [Google Scholar] [CrossRef]
- Hamman, J.H.; Enslin, G.M.; Kotzé, A.F. Oral Delivery of Peptide Drugs. BioDrugs 2005, 19, 165–177. [Google Scholar] [CrossRef]
- Cleland, J.L.; Daugherty, A.; Mrsny, R. Emerging Protein Delivery Methods. Curr. Opin. Biotechnol. 2001, 12, 212–219. [Google Scholar] [CrossRef]
- Bruckdorfer, T.; Marder, O.; Albericio, F. From Production of Peptides in Milligram Amounts for Research to Multi-tons Quantities for Drugs of the Future. Curr. Pharm. Biotechnol. 2004, 5, 29–43. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar]
- Enayat, S.; Ravanbod, S.; Rassoulzadegan, M.; Jazebi, M.; Tarighat, S.; Ala, F.; Emsley, J.; Othman, M. A novel D235Y mutation in the GP1BA gene enhances platelet interaction with von Willebrand factor in an Iranian family with platelet-type von Willebrand disease. Thomb. Haemost. 2012, 3, 946–954. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kazmirchuk, T.D.D.; Bradbury-Jost, C.; Withey, T.A.; Gessese, T.; Azad, T.; Samanfar, B.; Dehne, F.; Golshani, A. Peptides of a Feather: How Computation Is Taking Peptide Therapeutics under Its Wing. Genes 2023, 14, 1194. https://doi.org/10.3390/genes14061194
Kazmirchuk TDD, Bradbury-Jost C, Withey TA, Gessese T, Azad T, Samanfar B, Dehne F, Golshani A. Peptides of a Feather: How Computation Is Taking Peptide Therapeutics under Its Wing. Genes. 2023; 14(6):1194. https://doi.org/10.3390/genes14061194
Chicago/Turabian StyleKazmirchuk, Thomas David Daniel, Calvin Bradbury-Jost, Taylor Ann Withey, Tadesse Gessese, Taha Azad, Bahram Samanfar, Frank Dehne, and Ashkan Golshani. 2023. "Peptides of a Feather: How Computation Is Taking Peptide Therapeutics under Its Wing" Genes 14, no. 6: 1194. https://doi.org/10.3390/genes14061194