
Characterization and Phylogenetic Analysis of the Complete Mitochondrial Genome of Aythya marila
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics
2.2. Specimen Collection, Genome Sequencing, and Assembly
2.3. Annotation and Sequence Analysis
2.4. Phylogenetic Analysis
3. Results and Discussion
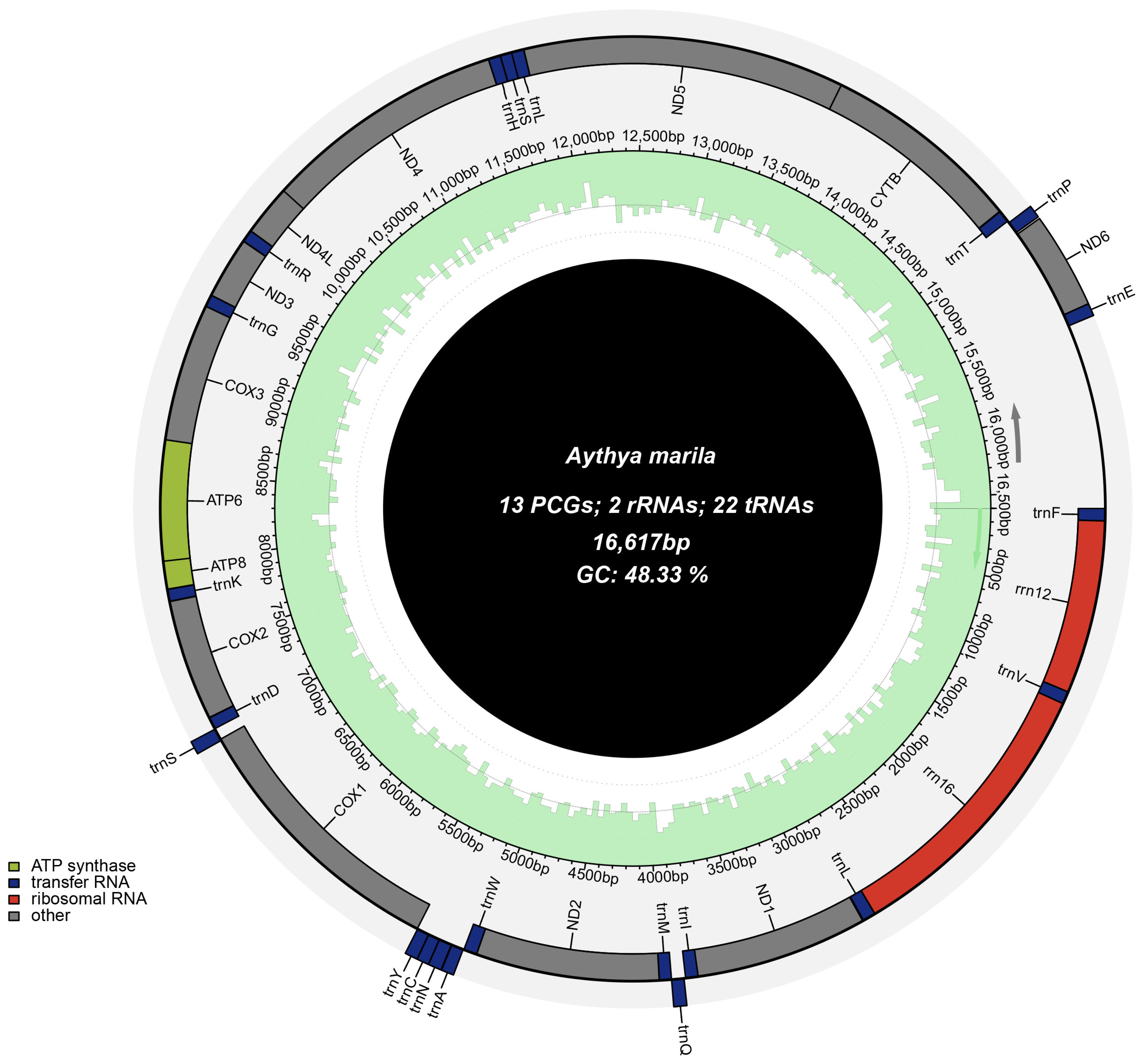
3.1. Genome Structure and Composition
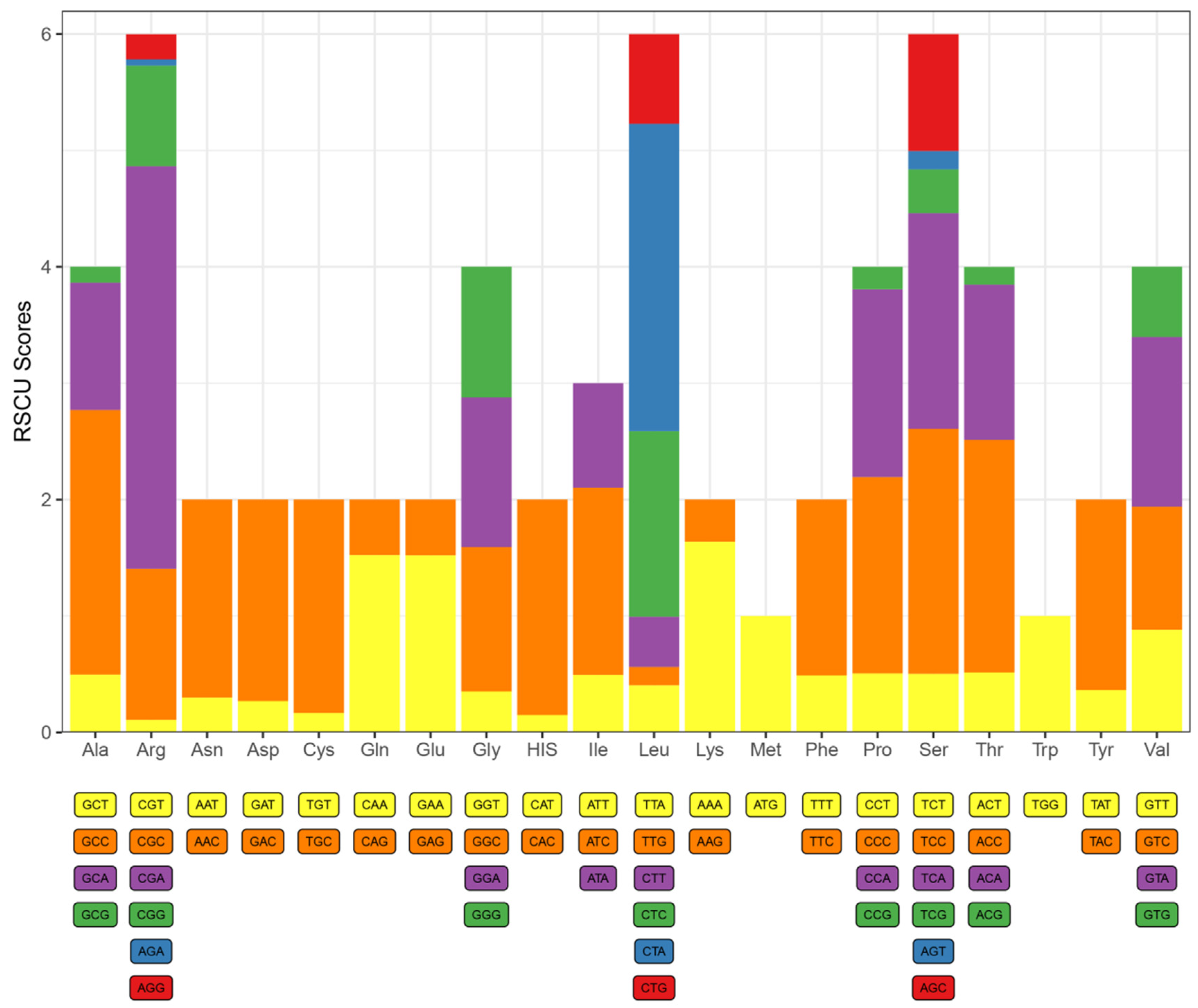
3.2. Protein-Coding Genes and Codon Usage
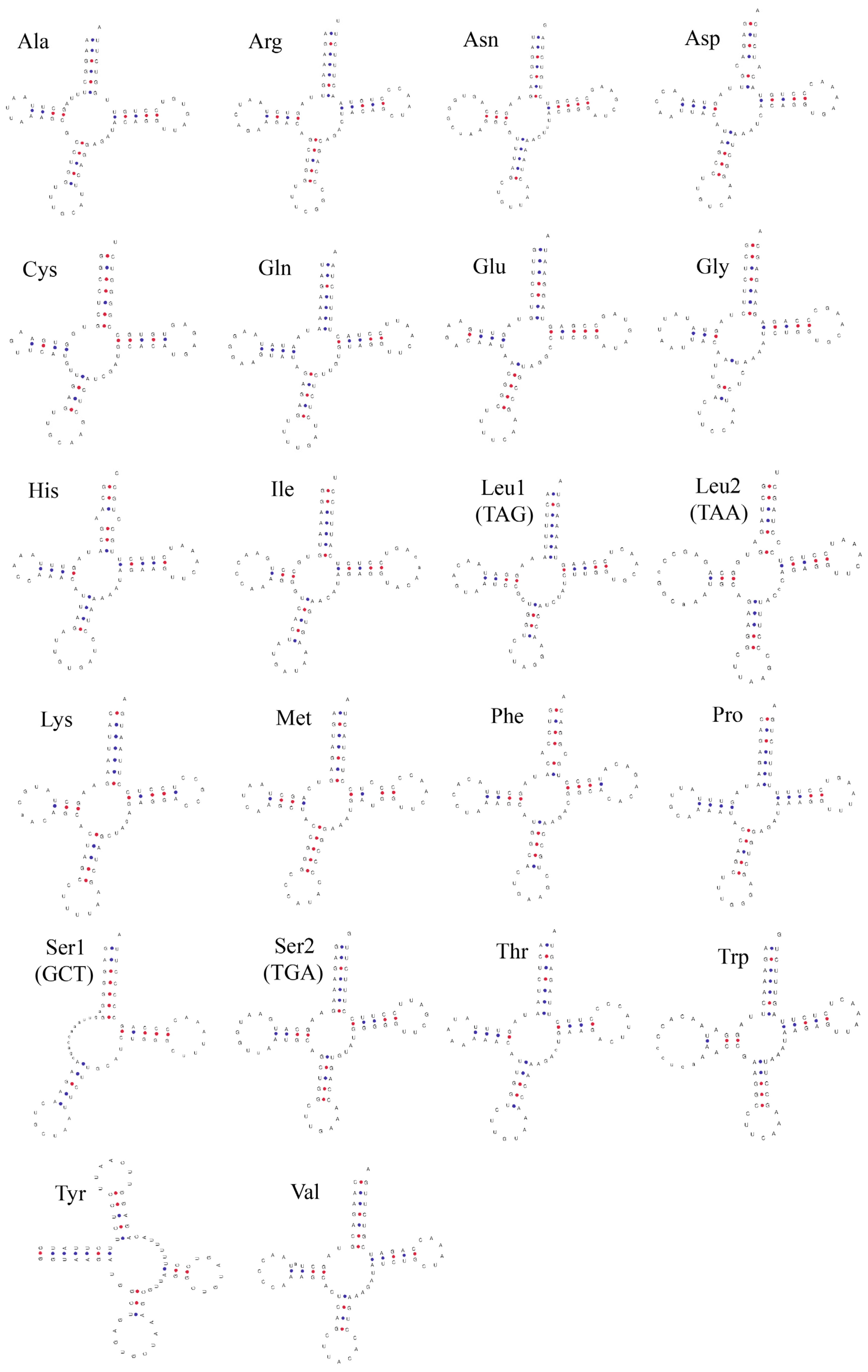
3.3. Ribosomal and Transfer RNA Genes
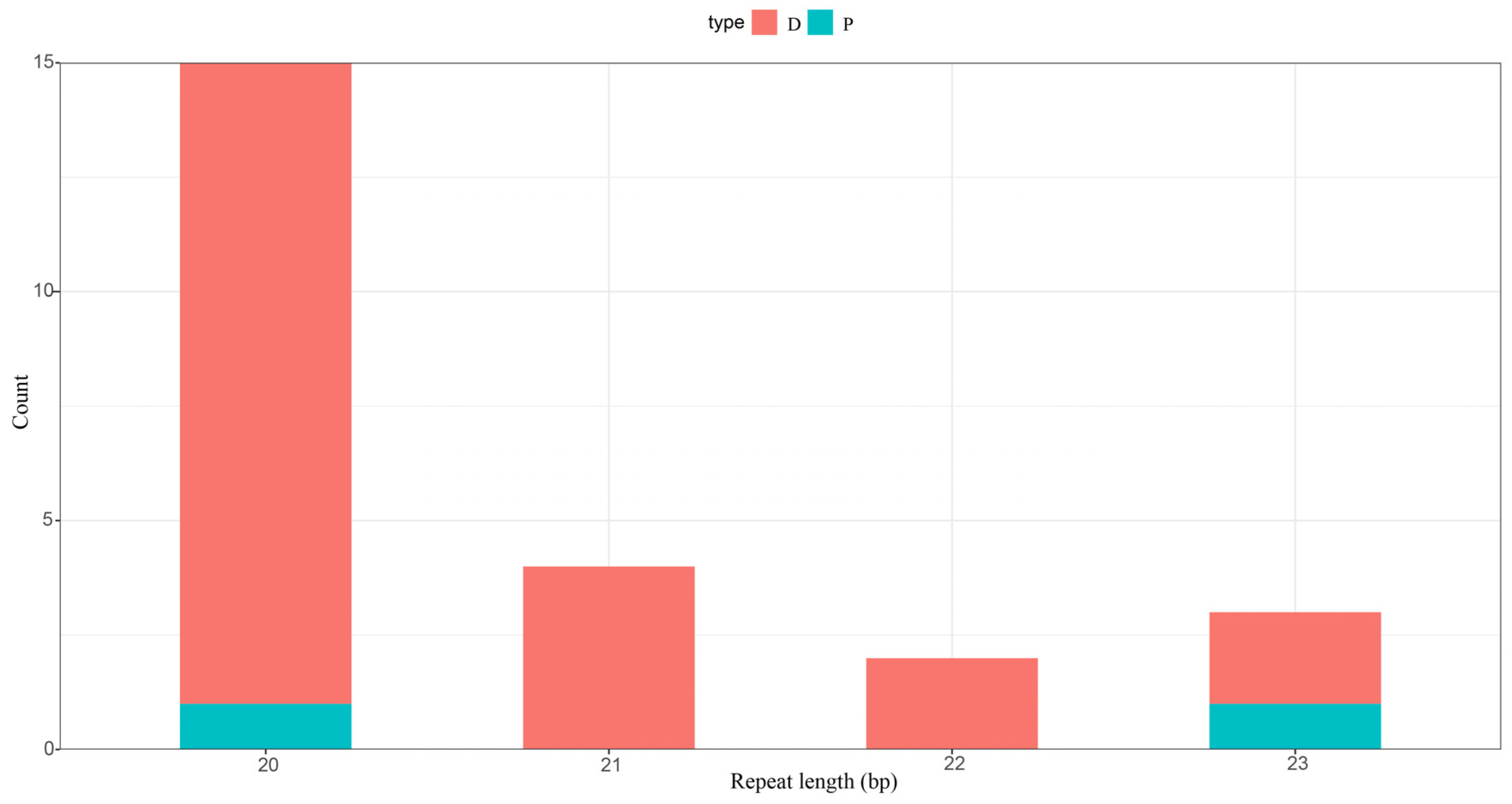
3.4. Control Region and Repetitive Sequences
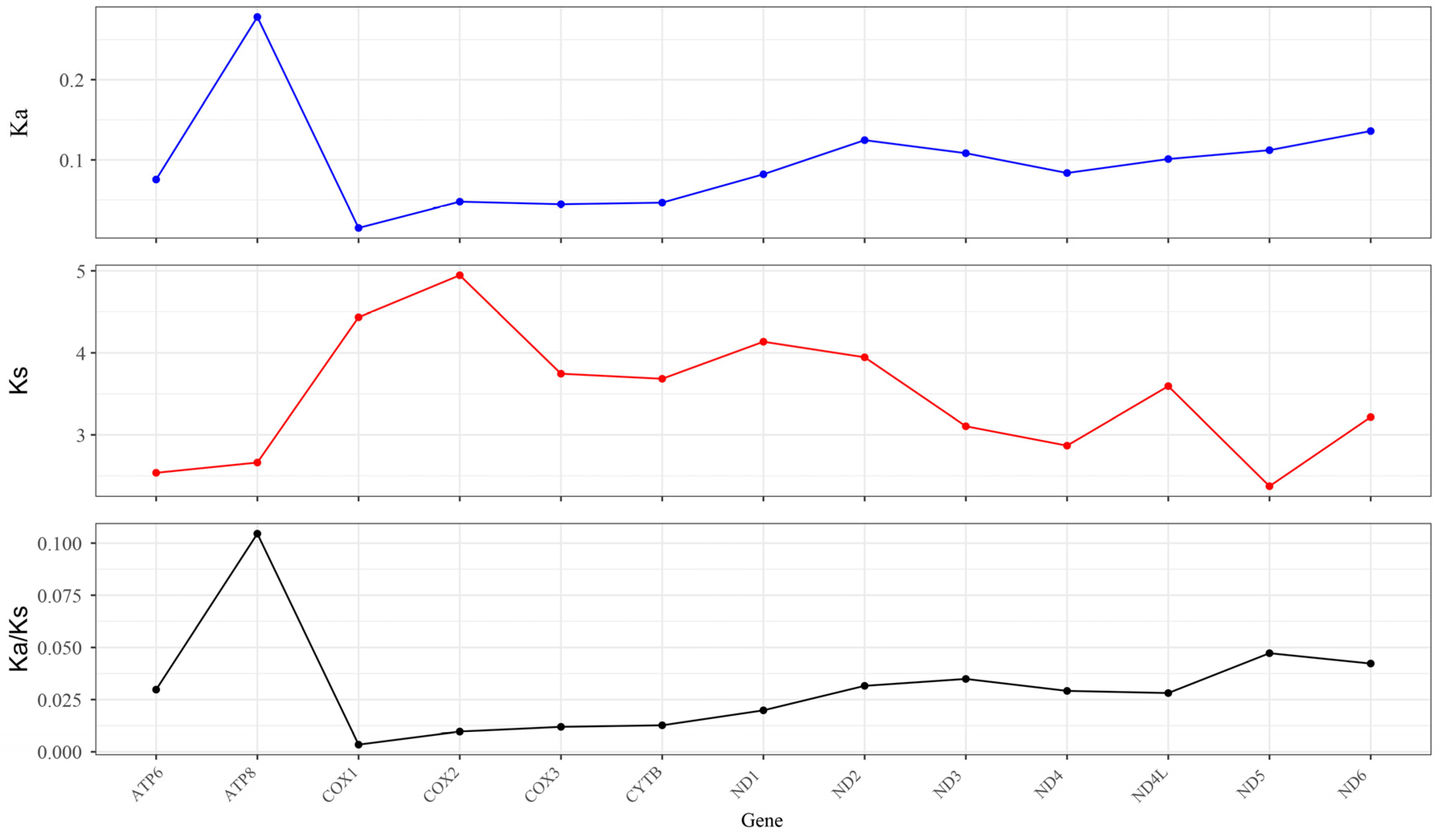
3.5. Synonymous (Ka) and Nonsynonymous (Ks) Substitution Rate
3.6. Population Genetic Diversity and Differentiation
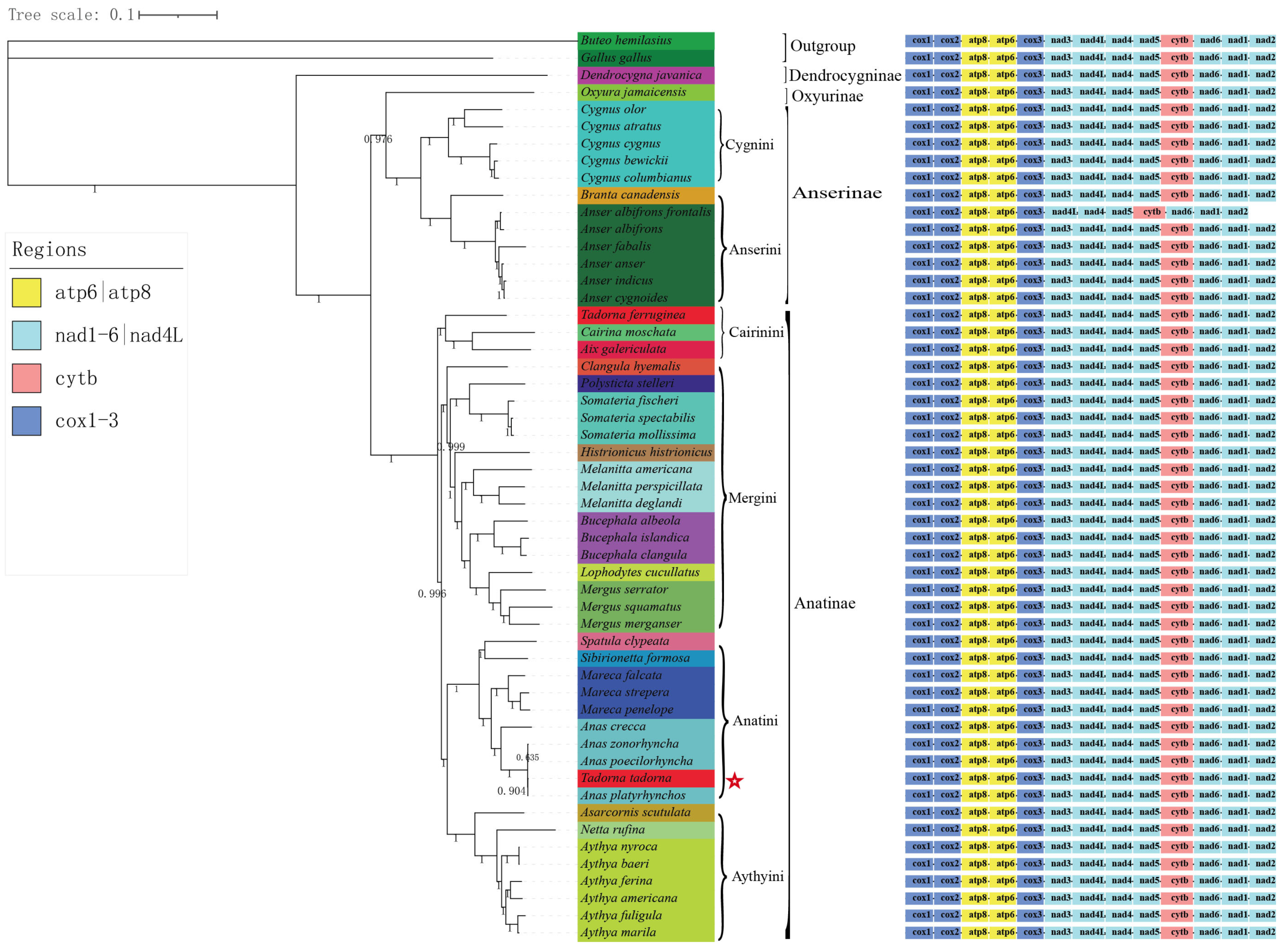
3.7. Phylogenetic Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Banks, R.C. Subspecies of the Greater Scaup and their names. Wilson Bull. 1986, 98, 433–444. [Google Scholar]
- Conover, H.B. Game birds of the Hooper Bay region, Alaska. Auk 1926, 43, 162–180. [Google Scholar] [CrossRef]
- Kessel, B.; Rocque, D.A.; Barclay, J.S. Greater Scaup (Aythya marila), version 1.0. In Birds of the World; Billerman, S.M., Ed.; Cornell Lab of Ornithology: Ithaca, NY, USA, 2020. [Google Scholar]
- Patton, J.; Avise, J. Evolutionary genetics of birds IV Rates of protein divergence in waterfowl (Anatidae). Genetica 1986, 68, 129–143. [Google Scholar] [CrossRef]
- Sun, Z.; Pan, T.; Hu, C.; Sun, L.; Ding, H.; Wang, H.; Zhang, C.; Jin, H.; Chang, Q.; Kan, X.; et al. Rapid and recent diversification patterns in Anseriformes birds: Inferred from molecular phylogeny and diversification analyses. PLoS ONE 2017, 12, e0184529. [Google Scholar] [CrossRef]
- Ottenburghs, J.; van Hooft, P.; van Wieren, S.E.; Ydenberg, R.C.; Prins, H.H. Hybridization in geese: A review. Front. Zool. 2016, 13, 20. [Google Scholar] [CrossRef]
- McCarthy, E.M. Handbook of Avian Hybrids of the World; Oxford University Press: New York, NY, USA, 2006. [Google Scholar]
- Van der Bliek, A.M.; Sedensky, M.M.; Morgan, P.G. Cell biology of the mitochondrion. Genetics 2017, 207, 843–871. [Google Scholar] [CrossRef]
- Ruokonen, M.; Kvist, L. Structure and evolution of the avian mitochondrial control region. Mol. Phylogenet. Evol. 2002, 23, 422–432. [Google Scholar] [CrossRef]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta (BBA)-Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Gissi, C.; Iannelli, F.; Pesole, G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 2008, 101, 301–320. [Google Scholar] [CrossRef]
- Subramanian, S.; Denver, D.R.; Millar, C.D.; Heupink, T.; Aschrafi, A.; Emslie, S.D.; Baroni, C.; Lambert, D.M. High mitogenomic evolutionary rates and time dependency. Trends Genet. 2009, 25, 482–486. [Google Scholar] [CrossRef]
- Sato, M.; Sato, K. Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2013, 1833, 1979–1984. [Google Scholar] [CrossRef]
- Li, Q.; Wei, S.-J.; Tang, P.; Wu, Q.; Shi, M.; Sharkey, M.J.; Chen, X.-X. Multiple lines of evidence from mitochondrial genomes resolve phylogenetic relationships of parasitic wasps in Braconidae. Genome Biol. Evol. 2016, 8, 2651–2662. [Google Scholar] [CrossRef]
- Pan, T.; Sun, Z.; Lai, X.; Orozcoterwengel, P.; Yan, P.; Wu, G.; Wang, H.; Zhu, W.; Wu, X.; Zhang, B. Hidden species diversity in Pachyhynobius: A multiple approaches species delimitation with mitogenomes. Mol. Phylogenetics Evol. 2019, 137, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Thapa, A.; Fan, H.; Ma, T.; Wu, Q.; Ma, S.; Zhang, D.; Wang, B.; Li, M.; Yan, L. Genomic evidence for two phylogenetic species and long-term population bottlenecks in red pandas. Sci. Adv. 2020, 6, eaax5751. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36 (Suppl. S2), W181–W184. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.; Biosciences, I.; Carlsbad, C. BioEdit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Lobry, J.R. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 1996, 13, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.-I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhou, Y.; Fei, Y.; Xie, C.; Hou, S. Mitochondrial genome of the critically endangered Baer’s Pochard, Aythya baeri, and its phylogenetic relationship with other Anatidae species. Sci. Rep. 2021, 11, 24302. [Google Scholar] [CrossRef]
- Mindell, D.P.; Sorenson, M.D.; Dimcheff, D.E. Multiple independent origins of mitochondrial gene order in birds. Proc. Natl. Acad. Sci. USA 1998, 95, 10693–10697. [Google Scholar] [CrossRef]
- Slack, K.E.; Janke, A.; Penny, D.; Arnason, U. Two new avian mitochondrial genomes (penguin and goose) and a summary of bird and reptile mitogenomic features. Gene 2003, 302, 43–52. [Google Scholar] [CrossRef]
- Hassanin, A.; Leger, N.; Deutsch, J. Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst. Biol. 2005, 54, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.L. Mitochondrial genome organization and vertebrate phylogenetics. Genet. Mol. Biol. 2000, 23, 745–752. [Google Scholar] [CrossRef]
- Steinberg, S.; Cedergren, R. Structural compensation in atypical mitochondrial tRNAs. Nat. Struct. Biol. 1994, 1, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Fourdrilis, S.; de Frias Martins, A.M.; Backeljau, T. Relation between mitochondrial DNA hyperdiversity, mutation rate and mitochondrial genome evolution in Melarhaphe neritoides (Gastropoda: Littorinidae) and other Caenogastropoda. Sci. Rep. 2018, 8, 17964. [Google Scholar] [CrossRef]
- Watanabe, Y.-I.; Suematsu, T.; Ohtsuki, T. Losing the stem-loop structure from metazoan mitochondrial tRNAs and co-evolution of interacting factors. Front. Genet. 2014, 5, 109. [Google Scholar] [CrossRef]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. TIG 2002, 18, 486. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Bielawski, J.P. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Lu, N.; Xu, Y.; He, C.; Lu, Z. Characterization and analysis of the mitochondrial genome of common bean (Phaseolus vulgaris) by comparative genomic approaches. Int. J. Mol. Sci. 2020, 21, 3778. [Google Scholar] [CrossRef]
- Delacour, J. Waterfowl of the World; Country Life Ltd.: London, UK, 1973. [Google Scholar]
- Livezey, B. A phylogenetic analysis of the whistling and white-backed ducks (Anatidae, Dendrocygninae) using morphological characters. Ann. Carnegie Mus. 1995, 64, 65–97. [Google Scholar] [CrossRef]
- Liu, G.; Zhou, L.; Li, B.; Zhang, L. The complete mitochondrial genome of Aix galericulata and Tadorna ferruginea: Bearings on their phylogenetic position in the Anseriformes. PLoS ONE 2014, 9, e109701. [Google Scholar] [CrossRef]
- Watanabe, J.; Matsuoka, H. Flightless diving duck (Aves, Anatidae) from the Pleistocene of Shiriya, northeast Japan. J. Vertebr. Paleontol. 2015, 35, e994745. [Google Scholar] [CrossRef]
- Sraml, M.; Christidis, L.; Easteal, S.; Horn, P.; Collet, C. Molecular relationships within Australasian waterfowl (Anseriformes). Aust. J. Zool. 1996, 44, 47–58. [Google Scholar] [CrossRef]
- Livezey, B.C. A phylogenetic analysis of geese and swans (Anseriformes: Anserinae), including selected fossil species. Syst. Biol. 1996, 45, 415–450. [Google Scholar] [CrossRef]
- Donne-Goussé, C.; Laudet, V.; Hänni, C. A molecular phylogeny of Anseriformes based on mitochondrial DNA analysis. Mol. Phylogenetics Evol. 2002, 23, 339–356. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Number | Sample Collection Site |
|---|---|---|
| A. marila | 6 | Heilongjiang Province, China |
| A. ferina | 6 | Heilongjiang Province, China |
| A. nyroca | 4 | Heilongjiang Province, China |
| A. baeri | 6 | Heilongjiang Province, China |
| A. fuligula | 6 | Heilongjiang Province, China |
| Gene | Strand | Location | Size (bp) | Intergenic Length | Anticodon | Start Codon | Stop Codon |
|---|---|---|---|---|---|---|---|
| tRNA-Phe | H | 1–70 | 70 | 0 | GAA | - | - |
| 12S rRNA | H | 71–1054 | 984 | 0 | - | - | - |
| tRNA-Val | H | 1055–1125 | 71 | 0 | TAC | - | - |
| 16S rRNA | H | 1126–2728 | 1603 | 0 | - | - | - |
| tRNA-Leu2 | H | 2729–2802 | 74 | 4 | TAA | - | - |
| ND1 | H | 2807–3784 | 978 | −2 | - | ATG | AGG |
| tRNA-Ile | H | 3783–3854 | 72 | 8 | GAT | - | - |
| tRNA-Gln | L | 3863–3933 | 71 | −1 | TGG | - | - |
| tRNA-Met | H | 3933–4001 | 69 | 0 | CAT | - | - |
| ND2 | H | 4002–5040 | 1039 | 0 | - | ATG | T-- |
| tRNA-Trp | H | 5041–5116 | 76 | 3 | TCA | - | - |
| tRNA-Ala | L | 5120–5188 | 69 | 2 | TGC | - | - |
| tRNA-Asn | L | 5191–5263 | 73 | 0 | GTT | - | - |
| tRNA-Cys | L | 5264–5329 | 66 | 0 | GCA | - | - |
| tRNA-Tyr | L | 5330–5399 | 70 | 1 | GTA | - | - |
| COI | H | 5401–6951 | 1551 | −9 | - | GTG | AGG |
| tRNA-Ser2 | L | 6943–7015 | 73 | 2 | TGA | - | - |
| tRNA-Asp | H | 7018–7086 | 69 | 1 | GTC | - | - |
| COII | H | 7088–7774 | 687 | 1 | - | GTG | TAA |
| tRNA-Lys | H | 7776–7843 | 68 | 1 | TTT | - | - |
| ATP8 | H | 7845–8012 | 168 | −10 | - | ATG | TAA |
| ATP6 | H | 8003–8686 | 684 | −1 | - | ATG | TAA |
| COIII | H | 8686–9469 | 784 | 0 | - | ATG | T-- |
| tRNA-Gly | H | 9470–9538 | 69 | 0 | TCC | - | - |
| ND3 | H | 9539–9890 | 351 | 1 | - | ATG | TAA |
| tRNA-Arg | H | 9892–9961 | 70 | 0 | TCG | - | - |
| ND4L | H | 9962–10,258 | 297 | −7 | - | ATG | TAA |
| ND4 | H | 10,252–11,629 | 1378 | 0 | - | ATG | T-- |
| tRNA-His | H | 11,630–11,698 | 69 | 0 | GTG | - | - |
| tRNA-Ser1 | H | 11,699–11,764 | 66 | −1 | GCT | - | - |
| tRNA-Leu1 | H | 11,764–11,834 | 71 | 0 | TAG | - | - |
| ND5 | H | 11,835–13,658 | 1824 | −1 | - | GTG | TAA |
| Cytb | H | 13,658–14,800 | 1143 | 2 | - | ATG | TAA |
| tRNA-Thr | H | 14,803–14,871 | 69 | 10 | TGT | - | - |
| tRNA-Pro | L | 14,882–14,951 | 70 | 10 | TGG | - | - |
| ND6 | L | 14,962–15,483 | 522 | 0 | - | ATG | TAG |
| tRNA-Glu | L | 15,484–15,551 | 68 | 0 | TTC | - | - |
| D-loop | H | 15,552–16,617 | 1066 | 0 | - | - | - |
| A. marila | T% | C% | A% | G% | (A + T)% | AT-Skew | GC-Skew | Length (bp) |
|---|---|---|---|---|---|---|---|---|
| Mitogenome | 22.25 | 32.84 | 29.42 | 15.49 | 51.67 | 0.139 | −0.359 | 16,617 |
| PCGs | 23.56 | 33.98 | 27.02 | 15.44 | 50.58 | 0.068 | −0.375 | 11,407 |
| COI | 24.11 | 32.95 | 25.98 | 16.96 | 50.09 | 0.037 | −0.320 | 1551 |
| COII | 22.12 | 33.19 | 27.66 | 17.03 | 49.75 | 0.111 | −0.315 | 687 |
| ATP8 | 17.86 | 40.47 | 33.93 | 7.74 | 51.79 | 0.310 | −0.679 | 168 |
| ATP6 | 23.39 | 37.99 | 27.92 | 11.70 | 51.31 | 0.088 | −0.540 | 684 |
| COIII | 23.09 | 33.67 | 26.91 | 16.33 | 50.00 | 0.076 | −0.347 | 784 |
| ND3 | 23.55 | 34.42 | 26.81 | 15.22 | 50.36 | 0.065 | −0.387 | 351 |
| ND5 | 21.32 | 35.09 | 29.99 | 13.60 | 51.31 | 0.169 | −0.441 | 1824 |
| ND4 | 22.71 | 36.00 | 29.32 | 11.97 | 52.03 | 0.127 | −0.501 | 1378 |
| ND4L | 24.58 | 33.67 | 25.59 | 16.16 | 50.17 | 0.020 | −0.351 | 297 |
| ND6 | 39.46 | 11.88 | 10.15 | 38.51 | 49.61 | −0.591 | 0.528 | 522 |
| Cytb | 23.01 | 36.31 | 26.33 | 14.35 | 49.34 | 0.067 | −0.433 | 1143 |
| ND1 | 24.34 | 34.66 | 25.36 | 15.64 | 49.70 | 0.021 | −0.378 | 978 |
| ND2 | 21.75 | 36.48 | 29.55 | 12.22 | 51.30 | 0.152 | −0.498 | 1039 |
| D-loop | 24.67 | 32.18 | 27.30 | 15.85 | 51.97 | 0.051 | −0.340 | 1066 |
| tRNAs | 26.64 | 22.03 | 30.01 | 21.32 | 56.65 | 0.059 | −0.016 | 1543 |
| 12S rRNA | 19.21 | 28.25 | 32.62 | 20.02 | 51.83 | 0.259 | −0.171 | 984 |
| 16S rRNA | 19.90 | 25.76 | 34.56 | 19.78 | 54.46 | 0.269 | −0.131 | 1603 |
| Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU(F) | 52 | 0.475 | UCU(S) | 23 | 0.491 | UAU(Y) | 18 | 0.340 | UGU(C) | 2 | 0.148 |
| UUC(F) | 167 | 1.525 | UCC(S) | 100 | 2.135 | UAC(Y) | 88 | 1.660 | UGC(C) | 27 | 1.852 |
| UUA(L) | 41 | 0.373 | UCA(S) | 87 | 1.858 | UAA(*) | 7 | - | UGA(*) | 87 | - |
| UUG(L) | 13 | 0.118 | UCG(S) | 13 | 0.278 | UAG(*) | 1 | - | UGG(W) | 19 | 1 |
| CUU(L) | 46 | 0.419 | CCU(P) | 25 | 0.435 | CAU(H) | 12 | 0.216 | CGU(R) | 1 | 0.081 |
| CUC(L) | 166 | 1.511 | CCC(P) | 94 | 1.635 | CAC(H) | 99 | 1.784 | CGC(R) | 16 | 0.297 |
| CUA(L) | 317 | 2.887 | CCA(P) | 102 | 1.774 | CAA(Q) | 67 | 1.489 | CGA(R) | 45 | 3.649 |
| CUG(L) | 76 | 0.692 | CCG(P) | 9 | 0.156 | CAG(Q) | 23 | 0.511 | CGG(R) | 10 | 0.811 |
| AUU(I) | 65 | 0.508 | ACU(T) | 31 | 0.392 | AAU(N) | 16 | 0.264 | AGU(S) | 7 | 0.241 |
| AUC(I) | 208 | 1.625 | ACC(T) | 156 | 1.975 | AAC(N) | 105 | 1.736 | AGC(S) | 51 | 1.759 |
| AUA(I) | 111 | 0.867 | ACA(T) | 115 | 1.456 | AAA(K) | 77 | 1.730 | AGA(R) | 0 | 0 |
| AUG(M) | 58 | 1 | ACG(T) | 14 | 0.177 | AAG(K) | 12 | 0.270 | AGG(R) | 2 | 0.162 |
| GUU(V) | 37 | 0.751 | GCU(A) | 40 | 0.559 | GAU(D) | 8 | 0.267 | GGU(G) | 11 | 0.204 |
| GUC(V) | 61 | 1.239 | GCC(A) | 173 | 2.420 | GAC(D) | 52 | 1.733 | GGC(G) | 77 | 1.426 |
| GUA(V) | 72 | 1.462 | GCA(A) | 91 | 1.273 | GAA(E) | 72 | 1.532 | GGA(G) | 74 | 1.370 |
| GUG(V) | 27 | 0.548 | GCG(A) | 12 | 0.168 | GAG(E) | 22 | 0.468 | GGG(G) | 54 | 1 |
| Species | NH | N | h | π |
|---|---|---|---|---|
| A. marila | 6 | 6 | 1 | 0.01109 |
| A. ferina | 6 | 6 | 1 | 0.00139 |
| A. nyroca | 4 | 4 | 1 | 0.00093 |
| A. baeri | 6 | 6 | 1 | 0.00080 |
| A. fuligula | 6 | 6 | 1 | 0.00087 |
| Species | A. fuligula | A. marila | A. ferina | A. nyroca |
|---|---|---|---|---|
| A. marila | 0.55425 | |||
| A. ferina | 0.96521 | 0.78815 | ||
| A. nyroca | 0.97498 | 0.82212 | 0.96901 | |
| A. baeri | 0.97628 | 0.82312 | 0.97023 | −0.09740 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Xia, T.; Gao, X.; Yang, X.; Sun, G.; Zhao, C.; Liu, G.; Zhang, H. Characterization and Phylogenetic Analysis of the Complete Mitochondrial Genome of Aythya marila. Genes 2023, 14, 1205. https://doi.org/10.3390/genes14061205
Zhang L, Xia T, Gao X, Yang X, Sun G, Zhao C, Liu G, Zhang H. Characterization and Phylogenetic Analysis of the Complete Mitochondrial Genome of Aythya marila. Genes. 2023; 14(6):1205. https://doi.org/10.3390/genes14061205
Chicago/Turabian StyleZhang, Lei, Tian Xia, Xiaodong Gao, Xiufeng Yang, Guolei Sun, Chao Zhao, Guangshuai Liu, and Honghai Zhang. 2023. "Characterization and Phylogenetic Analysis of the Complete Mitochondrial Genome of Aythya marila" Genes 14, no. 6: 1205. https://doi.org/10.3390/genes14061205