
Estimation of DNA Degradation in Archaeological Human Remains

 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Recovery of Ancient Samples
2.2. Sampling and Ethics Statement
2.3. DNA Extraction from Bone Findings
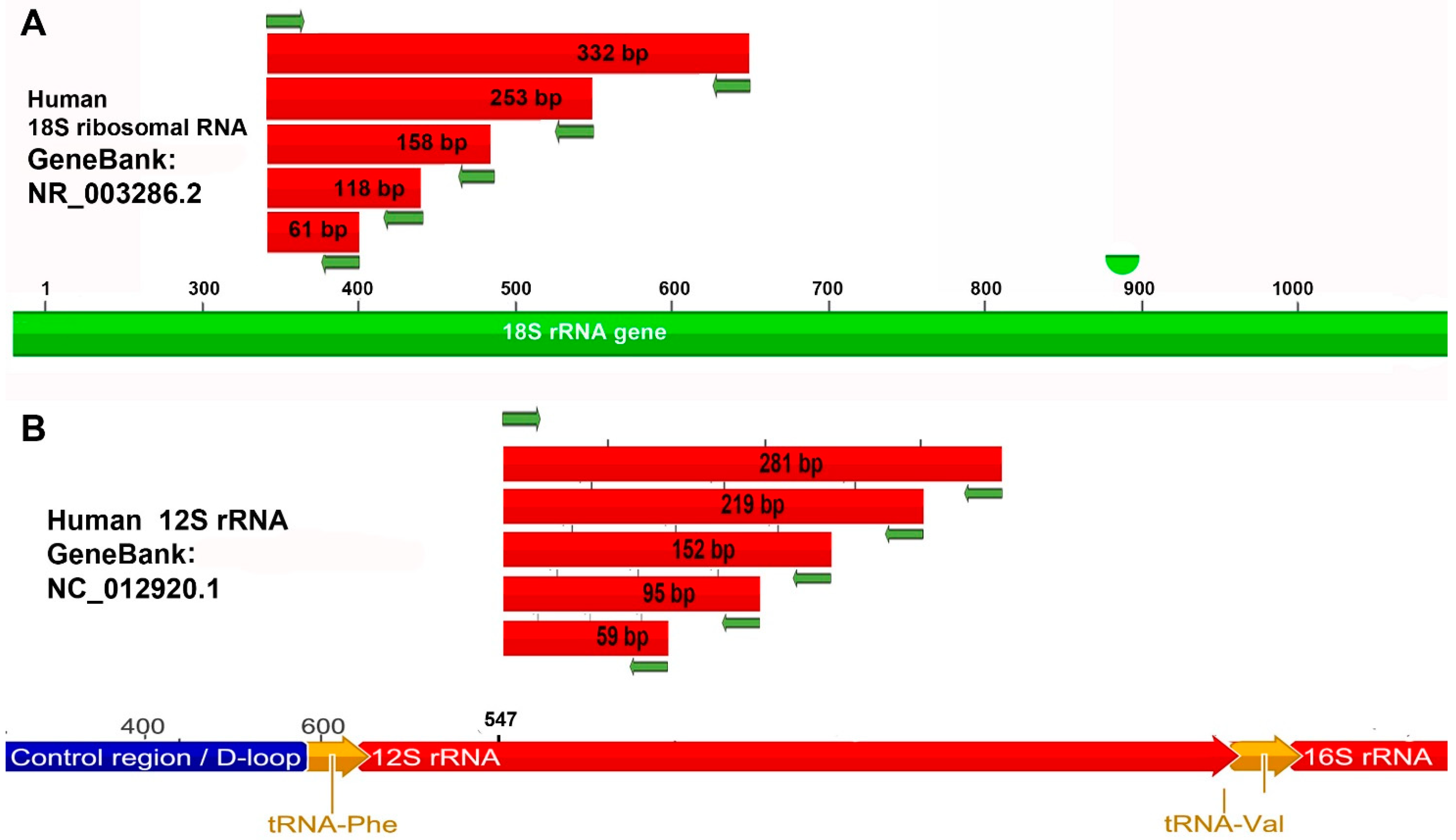
2.4. Primers: Design and Expected Amplicons
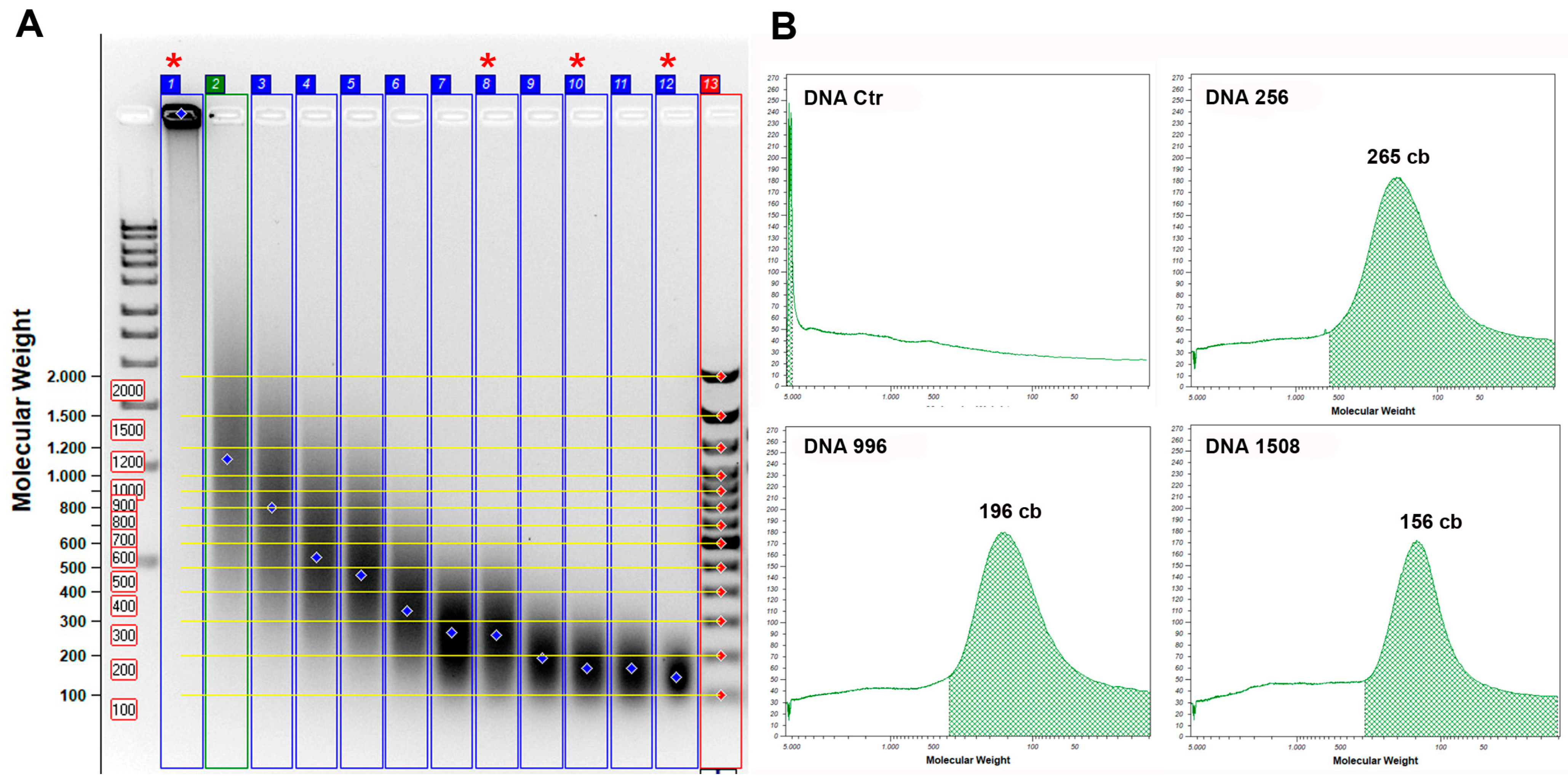
2.5. Standard DNA Fragmentation
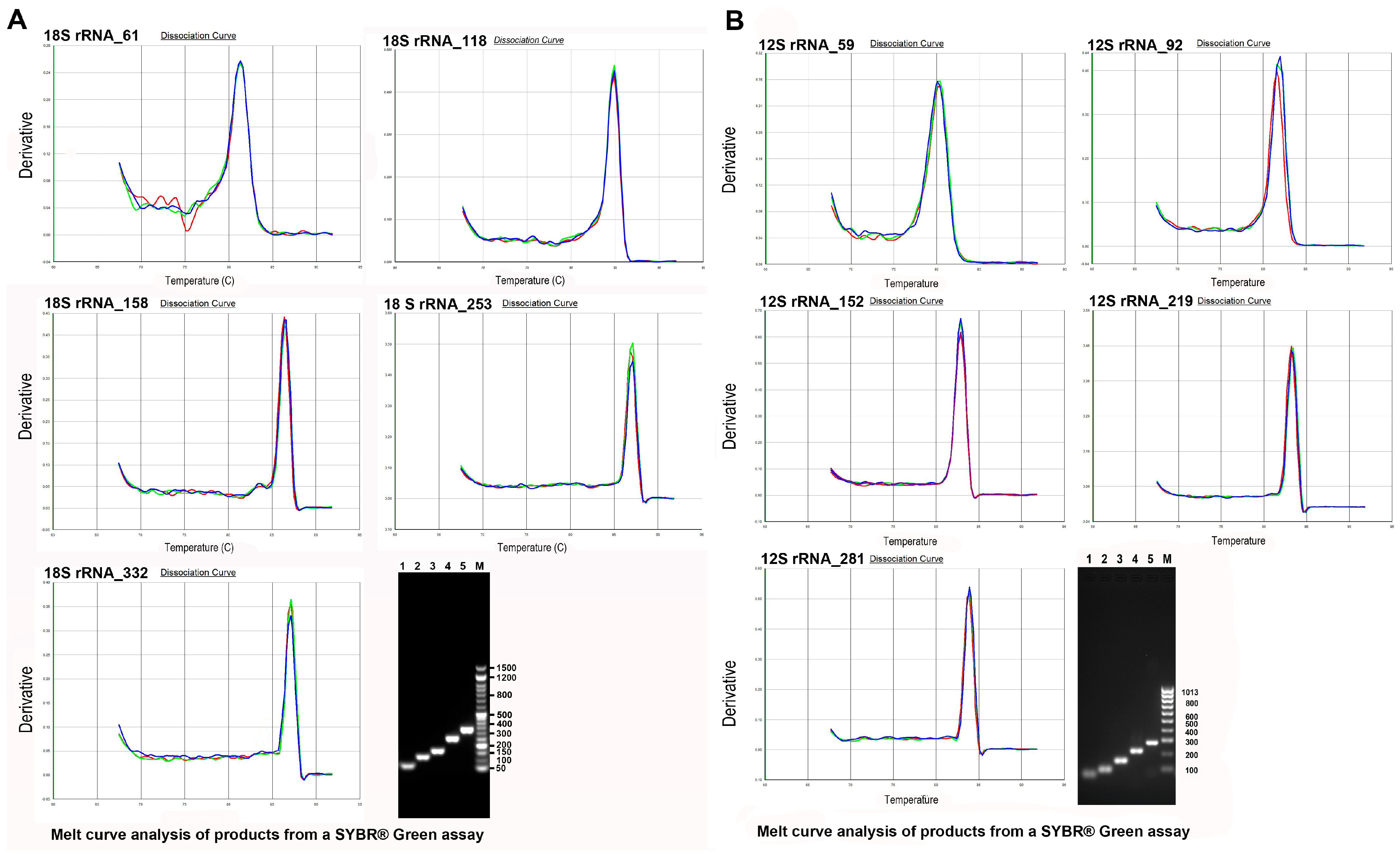
2.6. Standard Curves and Primer Efficiency
2.7. Quantitative Real-Time PCR and PCR
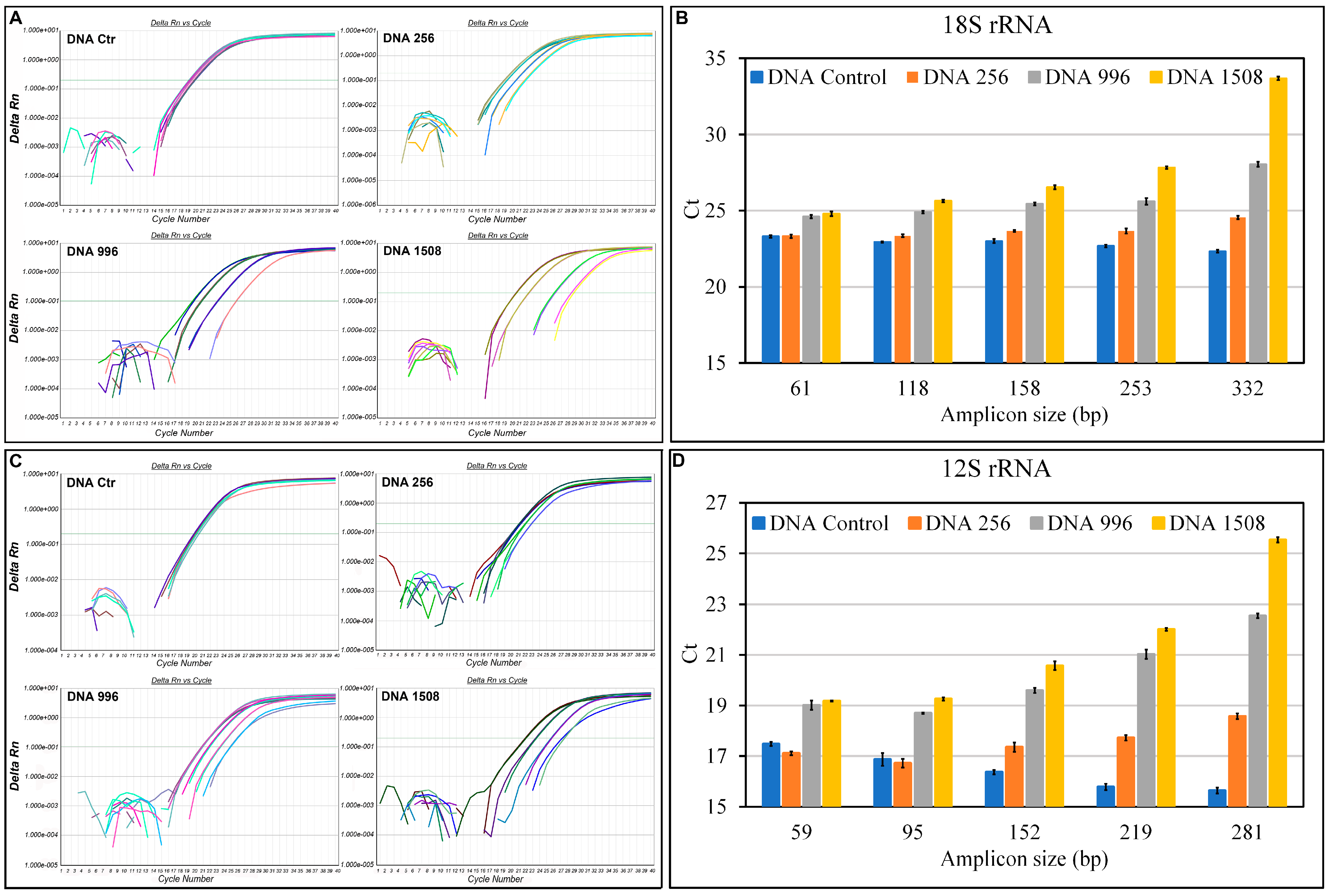
2.8. Quantification of aDNA Fragmentation
2.9. Electrophoresis
3. Results
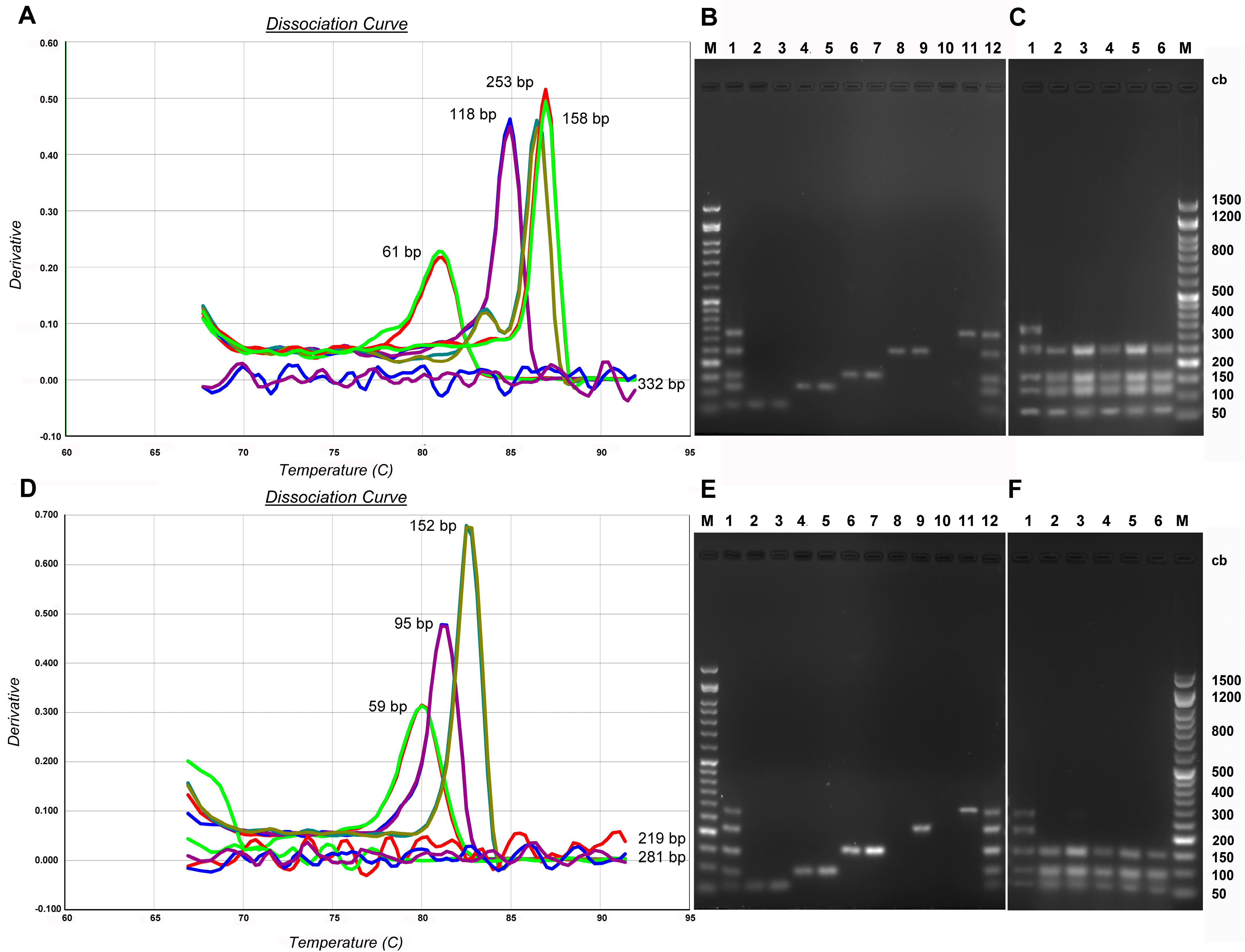
3.1. Standard DNA Fragmentation Analysis Subsection
3.2. Evaluation of Fragmentation in Standard DNA Subsubsection
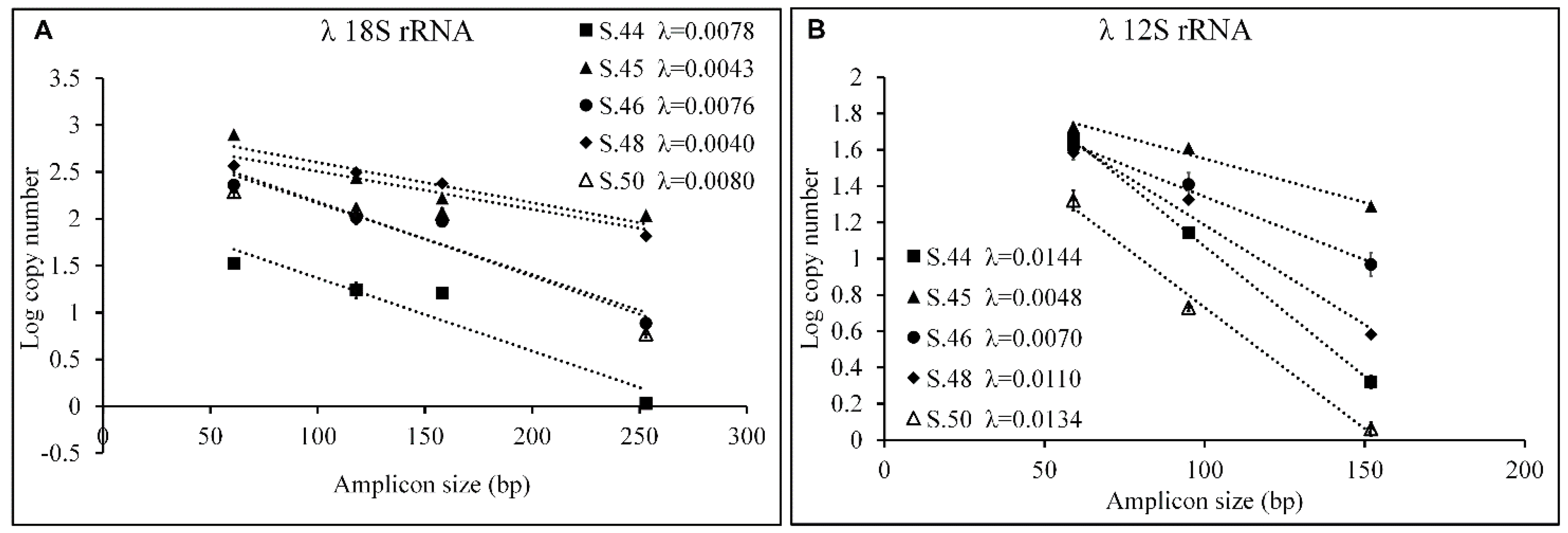
3.3. Evaluation of Fragmentation in aDNA
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adachi, N.; Shinoda, K.; Umetsu, K.; Matsumura, H. Mitochondrial DNA analysis of Jomon skeletons from the Funadomari site, Hokkaido, and its implication for the origins of Native American. Am. J. Phys. Anthropol. 2009, 138, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Anderson-Carpenter, L.L.; McLachlan, J.S.; Jackson, S.T.; Kuch, M.; Lumibao, C.Y.; Poinar, H.N. Ancient DNA from lake sediments: Bridging the gap between paleoecology and genetics. BMC Evol. Biol. 2011, 11, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, L.; Cooper, A. Using Ancient DNA to Understand Evolutionary and Ecological Processes. Annu. Rev. Ecol. Evol. Syst. 2014, 45, 573–598. [Google Scholar] [CrossRef]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.; Green, R.E.; Kelso, J.; Prüfer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pääbo, S.; Poinar, H.; Serre, D.; Jaenicke-Despres, V.; Hebler, J.; Rohland, N.; Kuch, M.; Krause, J.; Vigilant, L.; Hofreiter, M. Genetic analyses from ancient DNA. Annu. Rev. Genet. 2004, 38, 645–679. [Google Scholar] [CrossRef] [Green Version]
- Pilli, E.; Modi, A.; Serpico, C.; Achilli, A.; Lancioni, H.; Lippi, B.; Bertoldi, F.; Gelichi, S.; Lari, M.; Caramelli, D. Monitoring DNA contamination in handled vs. directly excavated ancient human skeletal remains. PLoS ONE 2013, 8, e52524. [Google Scholar] [CrossRef]
- Llamas, B.; Valverde, G.; Fehren-Schmitz, L.; Weyrich, L.S.; Cooper, A.; Haak, W. From the field to the laboratory: Controlling DNA contamination in human ancient DNA research in the high-throughput sequencing era. STAR Sci. Technol. Archaeol. Res. 2016, 3, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, T. Instability and Decay of the Primary Structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef]
- Pääbo, S. Ancient DNA: Extraction, characterization, molecular cloning, and enzymatic amplification. Proc. Natl. Acad. Sci. USA 1989, 86, 1939–1943. [Google Scholar] [CrossRef] [Green Version]
- Höss, M.; Jaruga, P.; Zastawny, T.H.; Dizdaroglu, M.; Pääbo, S. DNA damage and DNA sequence retrieval from ancient tissues. Nucleic Acids Res. 1996, 24, 1304–1307. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.; Willerslev, E.; Hansen, A. Damage and repair of ancient DNA. Mutat. Res. 2005, 571, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Zonta, E.; Nizard, P.; Taly, V. Assessment of DNA Integrity, Applications for Cancer Research. Adv. Clin. Chem. 2015, 70, 197–246. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, E.; Lari, M.; Gigli, E.; De Bellis, G.; Caramelli, D. Ancient DNA studies: New perspectives on old samples. Genet. Sel. Evol. 2012, 44, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pääbo, S. Amplifying ancient DNA. In PCR-Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 159–166. [Google Scholar]
- Poinar, H.; Kuch, M.; McDonald, G.; Martin, P.; Pääbo, S. Nuclear gene sequences from a late pleistocene sloth coprolite. Curr. Biol. 2003, 13, 1150–1152. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.T.P.; Hansen, A.J.; Willerslev, E.; Rudbeck, L.; Barnes, I.; Lynnerup, N.; Cooper, A. Characterization of genetic miscoding lesions caused by postmortem damage. Am. J. Hum. Genet. 2003, 72, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Lee, J.Y.; Bae, Y.K. Application of digital PCR for assessing DNA fragmentation in cytotoxicity response. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1235–1242. [Google Scholar] [CrossRef]
- Deagle, B.E.; Eveson, J.P.; Jarman, S.N. Quantification of damage in DNA recovered from highly degraded samples—A case study on DNA in faeces. Front. Zool. 2006, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Hashida, S.; Yamamoto, H.; Matsubara, T.; Ohtsuka, T.; Suzawa, K.; Maki, Y.; Soh, J.; Asano, H.; Tsukuda, K.; et al. Estimation of age-related DNA degradation from formalin-fixed and paraffin-embedded tissue according to the extraction methods. Exp. Ther. Med. 2017, 14, 2683–2688. [Google Scholar] [CrossRef] [Green Version]
- Robin, E.D.; Wong, R. Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. J. Cell. Physiol. 1988, 136, 507–513. [Google Scholar] [CrossRef]
- Thomas, R.H.; Schaffner, W.; Wilson, A.C.; Pääbo, S. DNA phylogeny of the extinct marsupial wolf. Nature 1989, 340, 465–467. [Google Scholar] [CrossRef]
- Cooper, A.; Lalueza-Fox, C.; Anderson, S.; Rambaut, A.; Austin, J.; Ward, R. Complete mitochondrial genome sequences of two extinct moas clarify ratite evolution. Nature 2001, 409, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, E.E.; James, H.F.; Olson, S.L.; Sorenson, M.D.; Jackson, J.; Fleischer, R.C. MtDNA from fossils reveals a radiation of Hawaiian geese recently derived from the Canada goose (Branta canadensis). Proc. Natl. Acad. Sci. USA 2002, 99, 1399–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hänni, C.; Laudet, V.; Stehelin, D.; Taberlet, P. Tracking the origins of the cave bear (Ursus spelaeus) by mitochondrial DNA sequencing. Proc. Natl. Acad. Sci. USA 1994, 91, 12336–12340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, L.; Calvignac, S.; Schnebelen, C.; Douady, C.J.; Godfrey, L.R.; Hänni, C. DNA from extinct giant lemurs links archaeolemurids to extant indriis. BMC Evol. Biol. 2008, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Driscoll, C.A.; Yamaguchi, N.; Bar-Gal, G.K.; Roca, A.L.; Luo, S.; Macdonald, D.W.; O’Brien, S.J. Mitochondrial phylogeography illuminates the origin of the extinct Caspian tiger and its relationship to the amur tiger. PLoS ONE 2009, 4, e4125. [Google Scholar] [CrossRef]
- Yang, L.; Tan, Z.; Wang, D.; Xue, L.; Guan, M.X.; Huang, T.; Li, R. Species identification through mitochondrial rRNA genetic analysis. Sci. Rep. 2014, 4, 4089. [Google Scholar] [CrossRef] [Green Version]
- Foran, D.R. Relative degradation of nuclear and mitochondrial DNA: An experimental approach. J. Forensic Sci. 2006, 51, 766–770. [Google Scholar] [CrossRef]
- Didelot, A.; Kotsopoulos, S.K.; Lupo, A.; Pekin, D.; Li, X.; Atochin, I.; Srinivasan, P.; Zhong, Q.; Olson, J.; Link, D.R.; et al. Multiplex picoliter-droplet digital PCR for quantitative assessment of DNA integrity in clinical samples. Clin. Chem. 2013, 59, 815–823. [Google Scholar] [CrossRef] [Green Version]
- Nicklas, J.A.; Buel, E. Development of an Alu-based, real-time PCR method for quantitation of human DNA in forensic samples. J. Forensic Sci. 2003, 48, 936–944. [Google Scholar] [CrossRef] [Green Version]
- Redi, F.; Forgione, A.; Savini, F.; Russi, A.; Siena, E.; De Iure, A. Amiternum (AQ): Scavo archeologico in località “Campo S. Maria”: Relazione preliminare, scavo 2013. Archeol. Mediev. 2014, 41, 171–194. [Google Scholar]
- Redi, F.; Forgione, A.; Siena, E.; Calabrese, L.; Ferretti, L. Amiternum (AQ): “Campo S. Maria”, campagna di scavo 2014. Archeol. Mediev. 2015, 42, 164–181. [Google Scholar]
- Fortea, J.; de la Rasila, M.; Garcı’a-Tabernero, A.; Gigli, E.; Rosas, A.; Lalueza-Fox, C. Excavation protocol of bone remains for Neandertal DNA analysis in El Sidrón Cave (Asturias, Spain). J. Hum. Evol. 2008, 55, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.; Cesare, P.; Bonfigli, A.; Volpe, A.R.; Colafarina, S.; Vecchiotti, G.; Forgione, A.; Zarivi, O. A qPCR-duplex assay for sex determination in ancient DNA. PLoS ONE 2022, 17, e0269913. [Google Scholar] [CrossRef] [PubMed]
- Redi, F.; Forgione, A.; Savini, F.; Siena, E.; De Iure, A.; Ciammetti, E. Amiternum (AQ): “Campo Santa Maria”, rapporto preliminare 2012. Archeol. Mediev. 2013, 40, 267–285. [Google Scholar]
- Gill, P.; Øyvind, B.; Fonneløp, A.E. Limitations of qPCR to estimate DNA quantity: An RFU method to facilitate inter-laboratory comparisons for activity level, and general applicability. Forensic Sci. Int. Genet. 2022, 61, 102777. [Google Scholar] [CrossRef]
- Dabney, J.; Meyer, M.; Pääbo, S. Ancient DNA damage. Cold Spring Harb. Perspect. Biol. 2013, 5, a012567. [Google Scholar] [CrossRef]
- Schwarz, C.; Debruyne, R.; Kuch, M.; McNally, E.; Schwarcz, H.; Aubrey, A.D.; Bada, J.; Poinar, H. New insights from old bones: DNA preservation and degradation in permafrost preserved mammoth remains. Nucleic Acids Res. 2009, 37, 3215–3229. [Google Scholar] [CrossRef]
- Götherström, A.; Collins, M.J.; Angerbjörn, A.; Lidén, K. Bone preservation and DNA amplification. Archaeometry 2002, 44, 395–404. [Google Scholar] [CrossRef]
- Allentoft, M.E.; Collins, M.; Harker, D.; Haile, J.; Oskam, C.L.; Hale, M.L.; Campos, P.F.; Samaniego, J.A.; Gilbert, M.T.; Willerslev, E.; et al. The half-life of DNA in bone: Measuring decay kinetics in 158 dated fossils. Proc. Biol. Sci. 2012, 279, 4724–4733. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, S.; Krause, J.; Guschanski, K.; Savolainen, V.; Pääbo, S. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS ONE 2012, 7, e34131. [Google Scholar] [CrossRef] [Green Version]
- Krause, J.; Fu, Q.; Good, J.M.; Viola, B.; Shunkov, M.V.; Derevianko, A.P.; Pääbo, S. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature 2010, 464, 894–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willerslev, E.; Cappellini, E.; Boomsma, W.; Nielsen, R.; Hebsgaard, M.B.; Brand, T.B.; Hofreiter, M.; Bunce, M.; Poinar, H.N.; Dahl-Jensen, D.; et al. Ancient biomolecules from deep ice cores reveal a forested southern Greenland. Science 2007, 317, 111–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez-García, T.A.; Vázquez-Domínguez, E.; Arroyo-Cabrales, J.; Kuch, M.; Enk, J.; King, C.; Poinar, H.N. Ancient DNA and the tropics: A rodent’s tale. Biol. Lett. 2014, 10, 20140224. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Bone | Age (Years) | Sex | Period |
|---|---|---|---|---|
| S.44 | Right femur | 45–50 (Adult) | Female | 9th–10th century |
| S.45 | Right femur | 45–50 (Adult) | Male | 9th–10th century |
| S.46 | Right femur | 16–18 (Teenager) | Male | 9th–10th century |
| S.48 | Left femur | 45–50 (Adult) | Male | 9th century |
| S.50 | Right femur | 45–50 (Adult) | Female | 12th century |
| Primer | Primer Sequence 5′-3′ | Tm | Product (bp) | Efficiency |
|---|---|---|---|---|
| F_18s-802 | GGCGGCTTTGGTGACTCTA | 58.8 °C | 802 | |
| R_18s-802 | TGGTCGGAACTACGACGGTAT | 59.8 °C | ||
| F_12s-1017 | TGTTTAGACGGGCTCACATCA | 57.9 °C | 2119 | |
| R_16S-61 | CTCCATAGGGTCTTCTCGTCTTG | 62.4 °C | ||
| F_ALU_50 R_ALU_50 | GATCACGAGGTCAGGAGGTC GGGTTTCACCGTTTTAGCCG | 61.4 °C 59.4 °C | 50 | 98% |
| F_18s | CGAACGTCTGCCCTATCAACTT | 60.3 °C | ||
| R_18s-61 | ACCCGTGGTCACCATGGTA | 58.8 °C | 61 | 100% |
| R_18s-118 | GGATGTGGTAGCCGTTTCTCA | 59.8 °C | 118 | 97% |
| R_18s-158 | GGGTCGGGAGTGGGTAATTT | 59.4 °C | 158 | 97% |
| R_18s-253 | CCAATGGATCCTCGTTAAAGGA | 58.4 °C | 253 | 101% |
| R_18s-332 | CGAGCTTTTTAACTGCAGCAACT | 58.9 °C | 332 | 95% |
| F_12s | AAAACTGCTCGCCAGAACACTAC | 60.6 °C | ||
| R_12s-59 | GCACCGCCAGGTCCTTT | 57.6 °C | 59 | 100% |
| R_12s-95 | TCGATTACAGAACAGGCTCCTCTA | 61.0 °C | 95 | 99% |
| R_12s-152 | TGCTGAAGATGGCGGTATATAGG | 60.6 °C | 152 | 97% |
| R_12s-219 | TGGGCTACACCTTGACCTAACG | 62.1 °C | 219 | 103% |
| R_12s-281 | TTTCATAAGGGCTATCGTAGTTTTCTG | 60.4 °C | 281 | 100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonfigli, A.; Cesare, P.; Volpe, A.R.; Colafarina, S.; Forgione, A.; Aloisi, M.; Zarivi, O.; Poma, A.M.G. Estimation of DNA Degradation in Archaeological Human Remains. Genes 2023, 14, 1238. https://doi.org/10.3390/genes14061238
Bonfigli A, Cesare P, Volpe AR, Colafarina S, Forgione A, Aloisi M, Zarivi O, Poma AMG. Estimation of DNA Degradation in Archaeological Human Remains. Genes. 2023; 14(6):1238. https://doi.org/10.3390/genes14061238
Chicago/Turabian StyleBonfigli, Antonella, Patrizia Cesare, Anna Rita Volpe, Sabrina Colafarina, Alfonso Forgione, Massimo Aloisi, Osvaldo Zarivi, and Anna Maria Giuseppina Poma. 2023. "Estimation of DNA Degradation in Archaeological Human Remains" Genes 14, no. 6: 1238. https://doi.org/10.3390/genes14061238
APA StyleBonfigli, A., Cesare, P., Volpe, A. R., Colafarina, S., Forgione, A., Aloisi, M., Zarivi, O., & Poma, A. M. G. (2023). Estimation of DNA Degradation in Archaeological Human Remains. Genes, 14(6), 1238. https://doi.org/10.3390/genes14061238