
Genomic Association Analysis of Growth and Backfat Traits in Large White Pigs

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Feeding of Experimental Animals
2.3. Animals and Phenotypes
2.4. Genotyping and Quality Control
2.5. Population Genome Analysis
2.6. Genome-Wide Analysis
2.7. Candidate Gene Annotation and Functional Enrichment Analysis
3. Results
3.1. Phenotypic Data and Population Structure Analysis
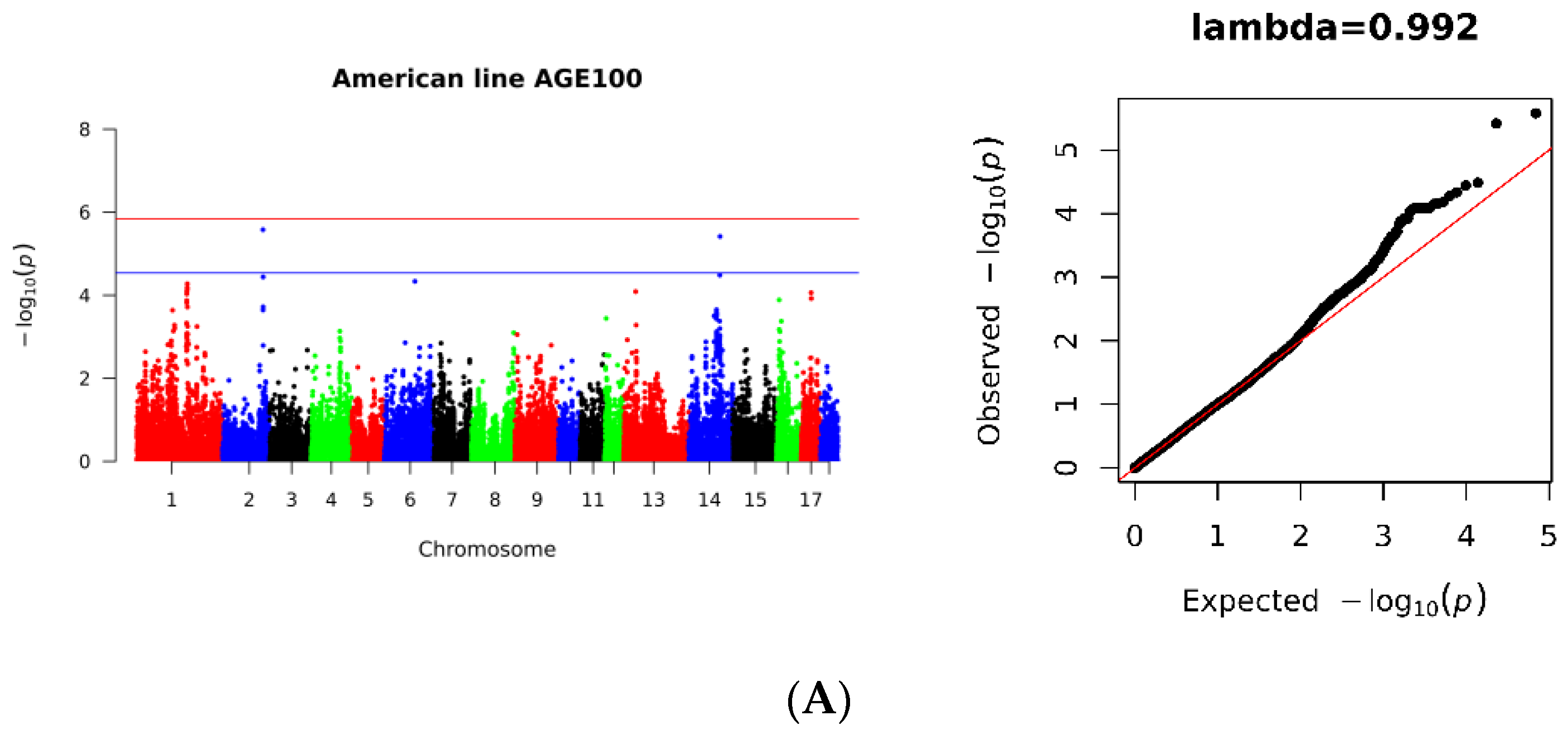
3.2. SNP Loci Associated with AGE100
3.3. SNPs Significantly Associated with BF100
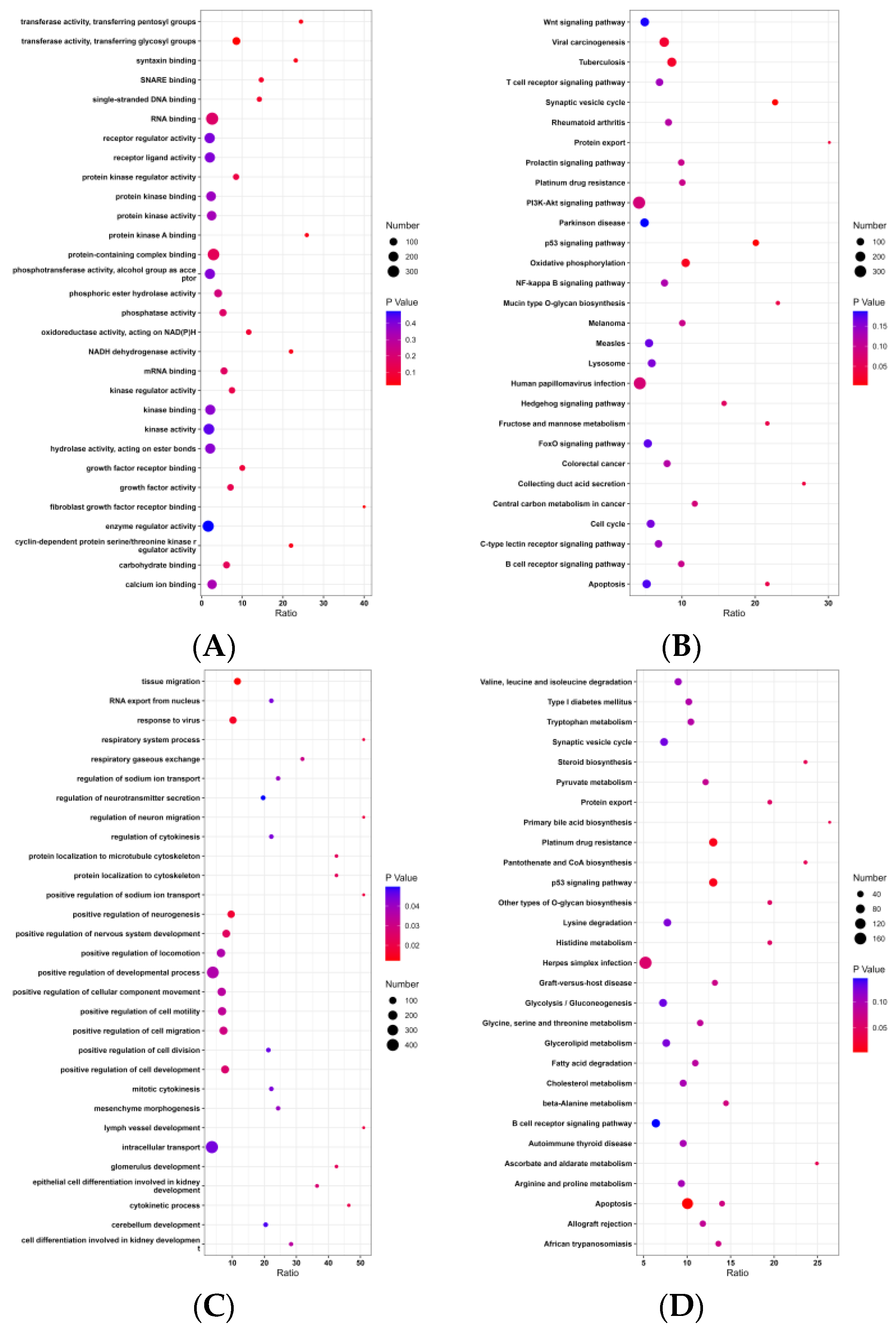
3.4. GO and KEGG Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, S.; Zhang, K.; Peng, X.; Zhan, H.; Lu, J.; Xie, S.; Zhao, S.; Li, X.; Ma, Y. Selective sweep analysis reveals extensive parallel selection traits between large white and Duroc pigs. Evol. Appl. 2020, 13, 2807–2820. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Z.B.; Nugent, R.A., 3rd. Heritability of body length and measures of body density and their relationship to backfat thickness and loin muscle area in swine. J. Anim. Sci. 2003, 81, 1943–1949. [Google Scholar] [CrossRef]
- Davoli, R.; Catillo, G.; Serra, A.; Zappaterra, M.; Zambonelli, P.; Zilio, D.M.; Steri, R.; Mele, M.; Buttazzoni, L.; Russo, V. Genetic parameters of backfat fatty acids and carcass traits in Large White pigs. Animal 2019, 13, 924–932. [Google Scholar] [CrossRef]
- Bidanel, J.P.; Ducos, A.; Gueblez, R.; Labroue, F. Genetic-parameters of bcakfat thickness, age at 100kg and ultimate pH in on-farm tested French Landrace and Large White pigs. Livest. Prod. Sci. 1994, 40, 291–301. [Google Scholar] [CrossRef]
- Ruan, D.; Zhuang, Z.; Ding, R.; Qiu, Y.; Zhou, S.; Wu, J.; Xu, C.; Hong, L.; Huang, S.; Zheng, E.; et al. Weighted single-step GWAS identified candidate genes associated with growth traits in a Duroc pig population. Genes 2021, 12, 117. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Tang, S.; Wang, C.; Wang, Y.; Qin, Y.; Wang, Y.; Zhang, J.; Song, H.; Mi, S.; Yu, F.; et al. A genome-wide association study of growth and fatness traits in two pig populations with different genetic backgrounds. J. Anim. Sci. 2018, 96, 806–816. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Gong, H.; Cui, L.; Zhang, W.; Ma, J.; Chen, C.; Ai, H.; Xiao, S.; Huang, L.; et al. Genetic correlation of fatty acid composition with growth, carcass, fat deposition and meat quality traits based on GWAS data in six pig populations. Meat Sci. 2019, 150, 47–55. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Shi, L.; Zhang, P.; Li, Y.; Li, M.; Tian, J.; Wang, L.; Zhao, F. GWAS of reproductive traits in Large White pigs on chip and imputed whole-genome sequencing data. Int. J. Mol. Sci. 2022, 23, 13338. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, X.; Tan, Z.; Ning, C.; Xing, K.; Yang, T.; Pan, Y.; Sun, D.; Wang, C. Genome-wide association study of piglet uniformity and farrowing interval. Front. Genet. 2017, 8, 194. [Google Scholar] [CrossRef]
- Okamura, T.; Onodera, W.; Tayama, T.; Kadowaki, H.; Kojima-Shibata, C.; Suzuki, E.; Uemoto, Y.; Mikawa, S.; Hayashi, T.; Awata, T.; et al. A genome-wide scan for quantitative trait loci affecting respiratory disease and immune capacity in Landrace pigs. Anim. Genet. 2012, 43, 721–729. [Google Scholar] [CrossRef]
- Uemoto, Y.; Ichinoseki, K.; Matsumoto, T.; Oka, N.; Takamori, H.; Kadowaki, H.; Kojima-Shibata, C.; Suzuki, E.; Okamura, T.; Aso, H.; et al. Genome-wide association studies for production, respiratory disease, and immune-related traits in Landrace pigs. Sci. Rep. 2021, 11, 15823. [Google Scholar] [CrossRef] [PubMed]
- Falker-Gieske, C.; Blaj, I.; Preuß, S.; Bennewitz, J.; Thaller, G.; Tetens, J. GWAS for meat and carcass traits using imputed sequence level genotypes in pooled F2-designs in pigs. G3 Genes Genomes Genet. 2019, 9, 2823–2834. [Google Scholar] [CrossRef] [PubMed]
- Oyelami, F.O.; Zhao, Q.; Xu, Z.; Zhang, Z.; Sun, H.; Zhang, Z.; Ma, P.; Wang, Q.; Pan, Y. Haplotype block analysis reveals candidate genes and QTLs for meat quality and disease resistance in Chinese Jiangquhai pig breed. Front. Genet. 2020, 11, 752. [Google Scholar] [CrossRef] [PubMed]
- Marigorta, U.M.; Rodríguez, J.A.; Gibson, G.; Navarro, A. Replicability and prediction: Lessons and challenges from GWAS. Trends Genet. 2018, 34, 504–517. [Google Scholar] [CrossRef]
- Colhoun, H.M.; McKeigue, P.M.; Davey Smith, G. Problems of reporting genetic associations with complex outcomes. Lancet 2003, 361, 865–872. [Google Scholar] [CrossRef]
- Evangelou, E.; Maraganore, D.M.; Ioannidis, J.P. Meta-analysis in genome-wide association datasets: Strategies and application in Parkinson disease. PLoS ONE 2007, 2, e196. [Google Scholar] [CrossRef]
- Wang, T. Genetic Parameter Estimates for the Age at 100 kg and Backfat Thickness in Large Whites. Master’s Thesis, China Agricultural University, Beijing, China, 2007. [Google Scholar]
- Chen, D.; Wu, P.; Yang, Q.; Wang, K.; Zhou, J.; Yang, X.; Jiang, A.; Shen, L.; Xiao, W.; Jiang, Y.; et al. Genome-wide association study for backfat thickness at 100 kg and loin muscle thickness in domestic pigs based on genotyping by sequencing. Physiol. Genom. 2019, 51, 261–266. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Li, Y.; Abecasis, G.R. METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010, 26, 2190–2191. [Google Scholar] [CrossRef]
- Wang, J.; Vasaikar, S.; Shi, Z.; Greer, M.; Zhang, B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017, 45, W130–W137. [Google Scholar] [CrossRef]
- Segarra, M.; Aburto, M.R.; Cop, F.; Llaó-Cid, C.; Härtl, R.; Damm, M.; Bethani, I.; Parrilla, M.; Husainie, D.; Schänzer, A.; et al. Endothelial Dab1 signaling orchestrates neuro-glia-vessel communication in the central nervous system. Science 2018, 361, eaao2861. [Google Scholar] [CrossRef] [PubMed]
- Rahimi-Balaei, M.; Bergen, H.; Kong, J.; Marzban, H. Neuronal migration during development of the Cerebellum. Front. Cell. Neurosci. 2018, 12, 484. [Google Scholar] [CrossRef] [PubMed]
- Smits, D.J.; Schot, R.; Wilke, M.; van Slegtenhorst, M.; de Wit, M.C.Y.; Dremmen, M.H.G.; Dobyns, W.B.; Barkovich, A.J.; Mancini, G.M.S. Biallelic DAB1 variants are associated with Mild Lissencephaly and Cerebellar Hypoplasia. Neurol. Genet. 2021, 7, e558. [Google Scholar] [CrossRef]
- Davisson, M.T.; Bronson, R.T.; Tadenev, A.L.; Motley, W.W.; Krishnaswamy, A.; Seburn, K.L.; Burgess, R.W. A spontaneous mutation in contactin 1 in the mouse. PLoS ONE 2011, 6, e29538. [Google Scholar] [CrossRef]
- Johansen, J.E.; Fetissov, S.O.; Bergström, U.; Nilsson, I.; Faÿ, C.; Ranscht, B.; Hökfelt, T.; Schalling, M. Evidence for hypothalamic dysregulation in mouse models of anorexia as well as in humans. Physiol. Behav. 2007, 92, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Xu, D.; Yang, C.; Wang, L.; Pan, W.; Zheng, C.; Fan, L. Contactin 1 as a potential biomarker promotes cell proliferation and invasion in thyroid cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 12473–12481. [Google Scholar] [PubMed]
- Chen, N.; He, S.; Geng, J.; Song, Z.-J.; Han, P.-H.; Qin, J.; Zhao, Z.; Song, Y.-C.; Wang, H.-X.; Dang, C.-X. Overexpression of Contactin 1 promotes growth, migration and invasion in Hs578T breast cancer cells. BMC Cell Biol. 2018, 19, 5. [Google Scholar] [CrossRef]
- Wang, B.B.; Hou, L.M.; Zhou, W.D.; Liu, H.; Tao, W.; Wu, W.J.; Niu, P.P.; Zhang, Z.P.; Zhou, J.; Li, Q.; et al. Genome-wide association study reveals a quantitative trait locus and two candidate genes on Sus scrofa chromosome 5 affecting intramuscular fat content in Suhuai pigs. Animal 2021, 15, 100341. [Google Scholar] [CrossRef]
- Yang, W.; Cao, H.; Xu, L.; Zhang, H.; Yan, Y. A novel eurythermic and thermostale lipase LipM from Pseudomonas moraviensis M9 and its application in the partial hydrolysis of algal oil. BMC Biotechnol. 2015, 15, 94. [Google Scholar] [CrossRef]
- Utsumi, T.; Hosokawa, T.; Shichita, M.; Nishiue, M.; Iwamoto, N.; Harada, H.; Kiwado, A.; Yano, M.; Otsuka, M.; Moriya, K. ANKRD22 is an N-myristoylated hairpin-like monotopic membrane protein specifically localized to lipid droplets. Sci. Rep. 2021, 11, 19233. [Google Scholar] [CrossRef]
- Balthasar, N.; Dalgaard, L.T.; Lee, C.E.; Yu, J.; Funahashi, H.; Williams, T.; Ferreira, M.; Tang, V.; McGovern, R.A.; Kenny, C.D.; et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 2005, 123, 493–505. [Google Scholar] [CrossRef]
- Onteru, S.K.; Gorbach, D.M.; Young, J.M.; Garrick, D.J.; Dekkers, J.C.; Rothschild, M.F. Whole genome association studies of residual feed intake and related traits in the pig. PLoS ONE 2013, 8, e61756. [Google Scholar] [CrossRef] [PubMed]
- Baron, M.; Maillet, J.; Huyvaert, M.; Dechaume, A.; Boutry, R.; Loiselle, H.; Durand, E.; Toussaint, B.; Vaillant, E.; Philippe, J.; et al. Loss-of-function mutations in MRAP2 are pathogenic in hyperphagic obesity with hyperglycemia and hypertension. Nat. Med. 2019, 25, 1733–1738. [Google Scholar] [CrossRef] [PubMed]
- Berruien, N.N.A.; Smith, C.L. Emerging roles of melanocortin receptor accessory proteins (MRAP and MRAP2) in physiology and pathophysiology. Gene 2020, 757, 144949. [Google Scholar] [CrossRef] [PubMed]
- Novoselova, T.V.; Larder, R.; Rimmington, D.; Lelliott, C.; Wynn, E.H.; Gorrigan, R.J.; Tate, P.H.; Guasti, L.; O’Rahilly, S.; Clark, A.J.L.; et al. Loss of Mrap2 is associated with Sim1 deficiency and increased circulating cholesterol. J. Endocrinol. 2016, 230, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Hua, H.; Xiao, G.; Yang, X.; Yang, Q.; Jin, L. ZC3HAV1 promotes the proliferation and metastasis via regulating KRAS in pancreatic cancer. Aging 2021, 13, 18482–18497. [Google Scholar] [CrossRef]
- Jiang, P.; Mao, L.; Lei, X.; Luo, C.; Zhang, Y.; Zhong, X.; Yin, Z.; Xu, X.; Li, D.; Zheng, Q. miR-1297 inhibits osteosarcoma cell proliferation and growth by targeting CCND2. Am. J. Cancer Res. 2022, 12, 3464–3478. [Google Scholar]
- Ficarelli, M.; Neil, S.J.D.; Swanson, C.M. Targeted restriction of viral gene expression and replication by the ZAP antiviral system. Annu. Rev. Virol. 2021, 8, 265–283. [Google Scholar] [CrossRef]
- Oliveira, H.C.; Derks, M.F.L.; Lopes, M.S.; Madsen, O.; Harlizius, B.; van Son, M.; Grindflek, E.H.; Gòdia, M.; Gjuvsland, A.B.; Otto, P.I.; et al. Fine mapping of a major backfat QTL reveals a causal regulatory variant affecting the CCND2 gene. Front. Genet. 2022, 13, 871516. [Google Scholar] [CrossRef] [PubMed]
- Le, T.H.; Christensen, O.F.; Nielsen, B.; Sahana, G. Genome-wide association study for conformation traits in three Danish pig breeds. Genet. Sel. Evol. 2017, 49, 12. [Google Scholar] [CrossRef] [PubMed]
- Suddason, T.; Gallagher, E. Genetic insights into Map3k-dependent proliferative expansion of T cells. Cell Cycle 2016, 15, 1956–1960. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | N | Day_Mean ± Std | bf_Mean ± Std | SNP_Num | After Filling SNP |
|---|---|---|---|---|---|
| Canadian line | 500 | 180.78 ± 17.57 | 12.14 ± 2.47 | 34,150 | 3,837,195 |
| Danish line | 314 | 167.29 ± 15.16 | 18.84 ± 4.4 | 34,543 | 12,437,523 |
| American line | 1500 | 161.48 ± 13.21 | 13.72 ± 2.77 | 34,497 | 8,077,147 |
| SSC (Sus scrofa Chromosome) | Position (bp) | p-Value | Distance | Genes |
|---|---|---|---|---|
| American line | ||||
| 14 | 101,189,804 | 3.83 × 10−6 | Upstream 463,324 bp | LIPM |
| 14 | 101,189,804 | 3.83 × 10−6 | Upstream 441,847 bp | ANKRD22 |
| Danish line | ||||
| 5 | 72,888,587 | 5.92 × 10−6 | Upstream 720,076 bp | CNTN1 |
| 5 | 72,888,587 | 5.92 × 10−6 | Within | PDZRN4 |
| Canadian line | ||||
| 6 | 154,765,159 | 5.96 × 10−7 | Downstream 664,281 bp | DAB1 |
| Meta-analysis | ||||
| 1 | 160,773,437 | 1.38 × 10−5 | Within | MC4R |
| 5 | 72,888,587 | 5.92 × 10−6 | Upstream 720,076 bp | CNTN1 |
| 5 | 72,888,587 | 5.92 × 10−6 | Within | PDZRN4 |
| 14 | 101,189,804 | 4.13 × 10−6 | Upstream 463,324 bp | LIPM |
| 14 | 101,189,804 | 4.13 × 10−6 | Upstream 441,847 bp | ANKRD22 |
| SSC (Sus scrofa Chormosome) | Position (bp) | p-Value | Distance | Gene |
|---|---|---|---|---|
| American line | ||||
| 16 | 35,401,235 | 4.77 × 10−6 | Downstream 470,895bp | MAP3K1 |
| Canadian line | ||||
| 1 | 53,461,852 | 6.19 × 10−6 | Upstream 205,846bp | MRAP2 |
| Meta-analysis | ||||
| 1 | 160,773,437 | 3.98 × 10−6 | Within | MC4R |
| 1 | 161,632,403 | 2.86 × 10−6 | Within | CPLX4 |
| 18 | 10,662,063 | 3.29 × 10−6 | Upstream 103,166bp | ZC3HAV1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, P.; Yin, C.; Wang, Y.; Yin, Z.; Liu, Y. Genomic Association Analysis of Growth and Backfat Traits in Large White Pigs. Genes 2023, 14, 1258. https://doi.org/10.3390/genes14061258
Zhou P, Yin C, Wang Y, Yin Z, Liu Y. Genomic Association Analysis of Growth and Backfat Traits in Large White Pigs. Genes. 2023; 14(6):1258. https://doi.org/10.3390/genes14061258
Chicago/Turabian StyleZhou, Peng, Chang Yin, Yuwei Wang, Zongjun Yin, and Yang Liu. 2023. "Genomic Association Analysis of Growth and Backfat Traits in Large White Pigs" Genes 14, no. 6: 1258. https://doi.org/10.3390/genes14061258
APA StyleZhou, P., Yin, C., Wang, Y., Yin, Z., & Liu, Y. (2023). Genomic Association Analysis of Growth and Backfat Traits in Large White Pigs. Genes, 14(6), 1258. https://doi.org/10.3390/genes14061258