
Leigh Syndrome Spectrum: A Portuguese Population Cohort in an Evolutionary Genetic Era

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
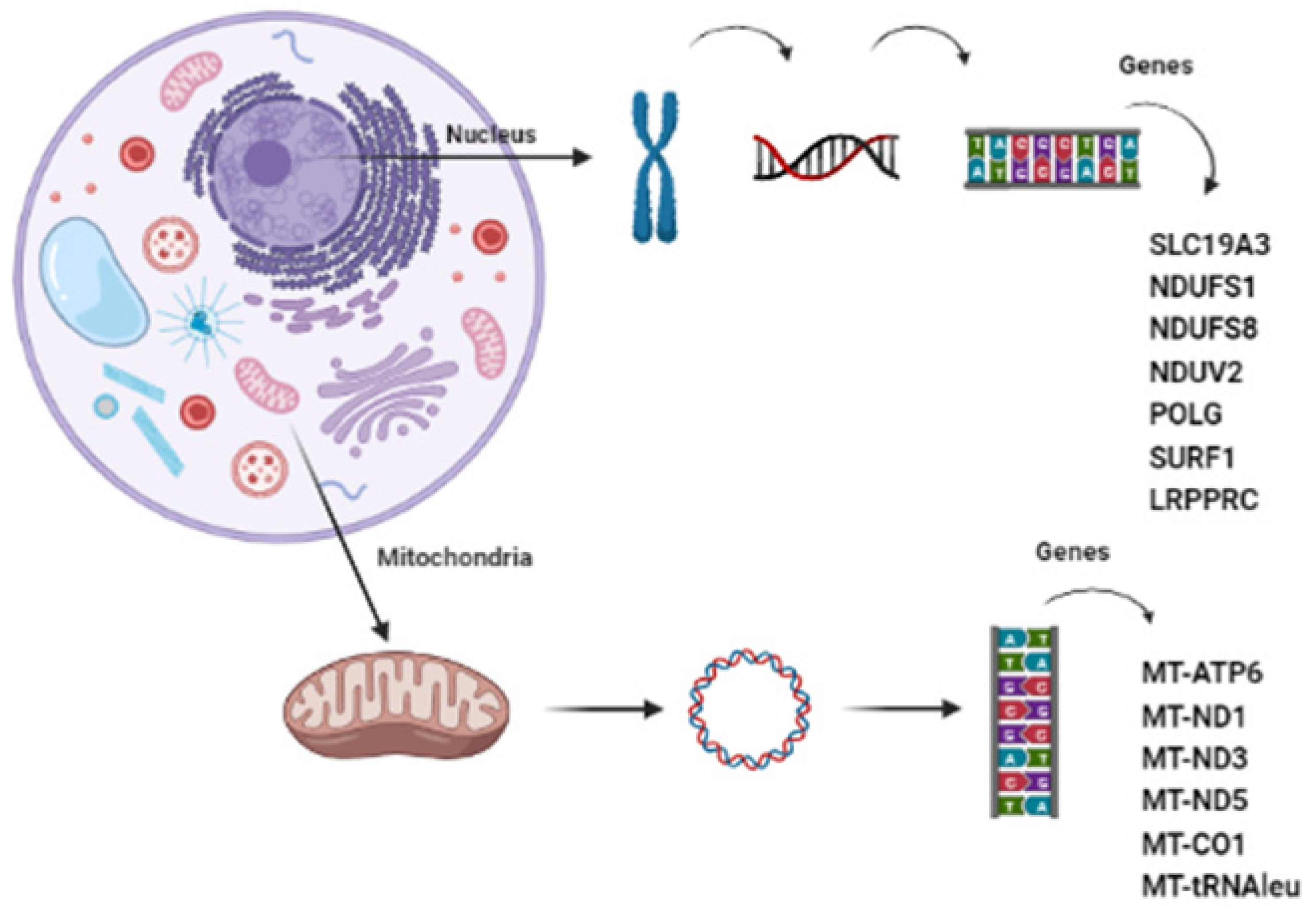
3.1. Mitochondrial DNA Mutations
3.2. Nuclear DNA Mutations
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
Nuclear Gene Panel for Mitochondrial Disorders
References
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh Syndrome: One Disorder, More than 75 Monogenic Causes: Leigh Syndrome. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef]
- Rahman, S.; Blok, R.B.; Dahl, H.-H.M.; Danks, D.M.; Kirby, D.M.; Chow, C.W.; Christodoulou, J.; Thorburn, D.R. Leigh Syndrome: Clinical Features and Biochemical and DNA Abnormalities. Ann. Neurol. 1996, 39, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Baertling, F.; Rodenburg, R.J.; Schaper, J.; Smeitink, J.A.; Koopman, W.J.H.; Mayatepek, E.; Morava, E.; Distelmaier, F. A Guide to Diagnosis and Treatment of Leigh Syndrome. J. Neurol. Neurosurg. Psychiatry 2014, 85, 257–265. [Google Scholar] [CrossRef]
- Holt, J.; Harding, A.E.; Petty, R.K.H.; Morgan-Hughes, J.A. A New Mitochondrial Disease Associated with Mitochondrial DNA Heteroplasmy. Am. J. Hum. Genet. 1990, 46, 428–433. [Google Scholar]
- Carrozzo, R.; Tessa, A.; Vazquez-Memije, M.E.; Piemonte, F.; Patrono, C.; Malandrini, A.; Dionisi-Vici, C.; Vilarinho, L.; Villanova, M.; Schagger, H.; et al. The T9176G MtDNA Mutation Severely Affects ATP Production and Results in Leigh Syndrome. Neurology 2001, 56, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Singh-Kler, R.; Hayes, C.M.; Smith, P.E.M.; Turnbull, D.M. Progressive Mitochondrial Disease Resulting from a Novel Missense Mutation in the Mitochondrial DNA ND3 Gene. Ann. Neurol. 2001, 50, 104–107. [Google Scholar] [CrossRef]
- Shoubridge, E.A. Nuclear Genetic Defects of Oxidative Phosphorylation. Hum. Mol. Genet. 2001, 10, 2277–2284. [Google Scholar] [CrossRef] [Green Version]
- Janssen, R.J.R.J.; Nijtmans, L.G.; Heuvel, L.P.V.D.; Smeitink, J.A.M. Mitochondrial Complex I: Structure, Function and Pathology. J. Inherit. Metab. Dis. 2006, 29, 499–515. [Google Scholar] [CrossRef]
- Rahman, S.; Thorburn, D. Nuclear Gene-Encoded Leigh Syndrome Spectrum Overview; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Thorburn, D.R.; Rahman, J.; Rahman, S. Mitochondrial DNA-Associated Leigh Syndrome and NARP. In Mitochondrial DNA; University of Washington: Seattle, WA, USA, 2017. [Google Scholar]
- Forny, P.; Footitt, E.; Davison, J.E.; Lam, A.; Woodward, C.E.; Batzios, S.; Bhate, S.; Chakrapani, A.; Cleary, M.; Gissen, P.; et al. Diagnosing Mitochondrial Disorders Remains Challenging in the Omics Era. Neurol. Genet. 2021, 7, e597. [Google Scholar] [CrossRef] [PubMed]
- Kremer, L.S.; Wortmann, S.B.; Prokisch, H. “Transcriptomics”: Molecular Diagnosis of Inborn Errors of Metabolism via RNA-Sequencing. J. Inherit. Metab. Dis. 2018, 41, 525–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenton, S.L.; Kremer, L.S.; Kopajtich, R.; Ludwig, C.; Prokisch, H. The Diagnosis of Inborn Errors of Metabolism by an Integrative “Multi-omics” Approach: A Perspective Encompassing Genomics, Transcriptomics, and Proteomics. J. Inherit. Metab. Dis. 2020, 43, 25–35. [Google Scholar] [CrossRef]
- Finsterer, J.; Laccone, F. Phenotypic Heterogeneity in 5 Family Members with the Mitochondrial Variant m.3243A>G. Am. J. Case Rep. 2020, 21, e927938. [Google Scholar] [CrossRef]
- Rahman, J.; Rahman, S. Mitochondrial Medicine in the Omics Era. Lancet 2018, 391, 2560–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakare, A.B.; Lesnefsky, E.J.; Iyer, S. Leigh Syndrome: A Tale of Two Genomes. Front. Physiol. 2021, 12, 693734. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the Effects of Coding Non-Synonymous Variants on Protein Function Using the SIFT Algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster Evaluates Disease-Causing Potential of Sequence Alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Povysil, G.; Tzika, A.; Vogt, J.; Haunschmid, V.; Messiaen, L.; Zschocke, J.; Klambauer, G.; Hochreiter, S.; Wimmer, K. Panelcn.MOPS: Copy-Number Detection in Targeted NGS Panel Data for Clinical Diagnostics. Hum. Mutat. 2017, 38, 889–897. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Cui, H.; Wong, L.-J.C. Comprehensive One-Step Molecular Analyses of Mitochondrial Genome by Massively Parallel Sequencing. Clin. Chem. 2012, 58, 1322–1331. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, C.; Silva, L.; Pereira, C.; Vieira, L.; Leão Teles, E.; Rodrigues, E.; Campos, T.; Janeiro, P.; Gaspar, A.; Dupont, J.; et al. Targeted next Generation Sequencing Identifies Novel Pathogenic Variants and Provides Molecular Diagnoses in a Cohort of Pediatric and Adult Patients with Unexplained Mitochondrial Dysfunction. Mitochondrion 2019, 47, 309–317. [Google Scholar] [CrossRef]
- De Vries, D.D.; Van Engelen, B.G.M.; Gabreëls, F.J.M.; Ruitenbeek, W.; Van Oost, B.A. A Second Missense Mutation in the Mitochondrial ATPase 6 Gene in Leigh’s Syndrome: MtDNA Mutation in Leigh’s Syndrome. Ann. Neurol. 1993, 34, 410–412. [Google Scholar] [CrossRef]
- Moslemi, A.-R.; Darin, N.; Tulinius, M.; Oldfors, A.; Holme, E. Two New Mutations in the MTATP6 Gene Associated with Leigh Syndrome. Neuropediatrics 2005, 36, 314–318. [Google Scholar] [CrossRef]
- Thyagarajan, D.; Shanske, S.; Vazquez-Memije, M.; Devivo, D.; Dimauro, S. A Novel Mitochondrial ATPase 6 Point Mutation in Familial Bilateral Striatal Necrosis. Ann. Neurol. 1995, 38, 468–472. [Google Scholar] [CrossRef]
- Poulton, J.; Turnbull, D.M.; Mehta, A.B.; Wilson, J.; Gardiner, R.M. Restriction Enzyme Analysis of the Mitochondrial Genome in Mitochondrial Myopathy. J. Med. Genet. 1988, 25, 600–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, D.M.; Salemi, R.; Sugiana, C.; Ohtake, A.; Parry, L.; Bell, K.M.; Kirk, E.P.; Boneh, A.; Taylor, R.W.; Dahl, H.-H.M.; et al. NDUFS6 Mutations Are a Novel Cause of Lethal Neonatal Mitochondrial Complex I Deficiency. J. Clin. Investig. 2004, 114, 837–845. [Google Scholar] [CrossRef]
- Valente, L.; Piga, D.; Lamantea, E.; Carrara, F.; Uziel, G.; Cudia, P.; Zani, A.; Farina, L.; Morandi, L.; Mora, M.; et al. Identification of Novel Mutations in Five Patients with Mitochondrial Encephalomyopathy. Biochim. Biophys. Acta BBA-Bioenerg. 2009, 1787, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Chol, M. The Mitochondrial DNA G13513A MELAS Mutation in the NADH Dehydrogenase 5 Gene Is a Frequent Cause of Leigh-like Syndrome with Isolated Complex I Deficiency. J. Med. Genet. 2003, 40, 188–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, C.; Souza, C.F.D.; Vedolin, L.; Vairo, F.; Lorea, C.; Sobreira, C.; Nogueira, C.; Vilarinho, L. Leigh Syndrome Due to MtDNA Pathogenic Variants. J. Inborn Errors Metab. Screen. 2019, 7, e2018000320. [Google Scholar] [CrossRef] [Green Version]
- Vilarinho, L.; Maia, C.; Coelho, T.; Coutinho, P.; Santorelli, F.M. Heterogeneous Presentation in Leigh Syndrome. J. Inherit. Metab. Dis. 1997, 20, 704–705. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Debs, R.; Depienne, C.; Rastetter, A.; Bellanger, A.; Degos, B.; Galanaud, D.; Keren, B.; Lyon-Caen, O.; Brice, A.; Sedel, F. Biotin-Responsive Basal Ganglia Disease in Ethnic Europeans with Novel SLC19A3 Mutations. Arch. Neurol. 2010, 67, 126–130. [Google Scholar] [CrossRef]
- Peters, H.; Buck, N.; Wanders, R.; Ruiter, J.; Waterham, H.; Koster, J.; Yaplito-Lee, J.; Ferdinandusse, S.; Pitt, J. ECHS1 Mutations in Leigh Disease: A New Inborn Error of Metabolism Affecting Valine Metabolism. Brain 2014, 137, 2903–2908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, H.; Ferdinandusse, S.; Ruiter, J.P.; Wanders, R.J.A.; Boneh, A.; Pitt, J. Metabolite Studies in HIBCH and ECHS1 Defects: Implications for Screening. Mol. Genet. Metab. 2015, 115, 168–173. [Google Scholar] [CrossRef]
- Bénit, P.; Beugnot, R.; Chretien, D.; Giurgea, I.; De Lonlay-Debeney, P.; Issartel, J.-P.; Corral-Debrinski, M.; Kerscher, S.; Rustin, P.; Rötig, A.; et al. Mutant NDUFV2 Subunit of Mitochondrial Complex I Causes Early Onset Hypertrophic Cardiomyopathy and Encephalopathy: NDUFV2 and cardiomyopathy/encephalopathy. Hum. Mutat. 2003, 21, 582–586. [Google Scholar] [CrossRef]
- Tiranti, V.; Hoertnagel, K.; Carrozzo, R.; Galimberti, C.; Munaro, M.; Granatiero, M.; Zelante, L.; Gasparini, P.; Marzella, R.; Rocchi, M.; et al. Mutations of SURF-1 in Leigh Disease Associated with Cytochrome c Oxidase Deficiency. Am. J. Hum. Genet. 1998, 63, 1609–1621. [Google Scholar] [CrossRef] [Green Version]
- Wojcik, M.H.; Schwartz, T.S.; Thiele, K.E.; Paterson, H.; Stadelmaier, R.; Mullen, T.E.; VanNoy, G.E.; Genetti, C.A.; Madden, J.A.; Gubbels, C.S.; et al. Infant Mortality: The Contribution of Genetic Disorders. J. Perinatol. 2019, 39, 1611–1619. [Google Scholar] [CrossRef]
- Ganetzky, R.D.; Stendel, C.; McCormick, E.M.; Zolkipli-Cunningham, Z.; Goldstein, A.C.; Klopstock, T.; Falk, M.J. MT-ATP6 Mitochondrial Disease Variants: Phenotypic and Biochemical Features Analysis in 218 Published Cases and Cohort of 14 New Cases. Hum. Mutat. 2019, 40, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.; Campos, Y.; Sánchez, J.M.; Bonaventura, I.; Aguilar, M.; García, A.; González, L.; Rey, M.J.; Arenas, J.; Olivé, M.; et al. NARP-MILS Syndrome Caused by 8993 T > G Mitochondrial DNA Mutation: A Clinical, Genetic and Neuropathological Study. Acta Neuropathol. 2006, 111, 610–616. [Google Scholar] [CrossRef]
- Craig, K.; Elliott, H.R.; Keers, S.M.; Lambert, C.; Pyle, A.; Graves, T.D.; Woodward, C.; Sweeney, M.G.; Davis, M.B.; Hanna, M.G.; et al. Episodic Ataxia and Hemiplegia Caused by the 8993T→C Mitochondrial DNA Mutation. J. Med. Genet. 2007, 44, 797–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramaniam, S.; Lewis, B.; Mock, D.M.; Said, H.M.; Tarailo-Graovac, M.; Mattman, A.; Van Karnebeek, C.D.; Thorburn, D.R.; Rodenburg, R.J.; Christodoulou, J. Leigh-Like Syndrome Due to Homoplasmic m.8993T>G Variant with Hypocitrullinemia and Unusual Biochemical Features Suggestive of Multiple Carboxylase Deficiency (MCD). In JIMD Reports; Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; Volume 33, pp. 99–107. [Google Scholar] [CrossRef] [Green Version]
- Debray, F.-G.; Lambert, M.; Allard, P.; Mitchell, G.A. Low Citrulline in Leigh Disease: Still a Biomarker of Maternally Inherited Leigh Syndrome. J. Child Neurol. 2010, 25, 1000–1002. [Google Scholar] [CrossRef] [PubMed]
- Peretz, R.H.; Ah Mew, N.; Vernon, H.J.; Ganetzky, R.D. Prospective Diagnosis of MT-ATP6-Related Mitochondrial Disease by Newborn Screening. Mol. Genet. Metab. 2021, 134, 37–42. [Google Scholar] [CrossRef]
- Tise, C.G.; Verscaj, C.P.; Mendelsohn, B.A.; Woods, J.; Lee, C.U.; Enns, G.M.; Stander, Z.; Hall, P.L.; Cowan, T.M.; Cusmano-Ozog, K.P. MT-ATP6 Mitochondrial Disease Identified by Newborn Screening Reveals a Distinct Biochemical Phenotype. Am. J. Med. Genet. A 2023, 191, 1492–1501. [Google Scholar] [CrossRef]
- Larson, A.A.; Balasubramaniam, S.; Christodoulou, J.; Burrage, L.C.; Marom, R.; Graham, B.H.; Diaz, G.A.; Glamuzina, E.; Hauser, N.; Heese, B.; et al. Biochemical Signatures Mimicking Multiple Carboxylase Deficiency in Children with Mutations in MT-ATP6. Mitochondrion 2019, 44, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Childs, A.-M.; Hutchin, T.; Pysden, K.; Highet, L.; Bamford, J.; Livingston, J.; Crow, Y. Variable Phenotype Including Leigh Syndrome with a 9185T>C Mutation in the MTATP6 Gene. Neuropediatrics 2007, 38, 313–316. [Google Scholar] [CrossRef]
- Pfeffer, G.; Blakely, E.L.; Alston, C.L.; Hassani, A.; Boggild, M.; Horvath, R.; Samuels, D.C.; Taylor, R.W.; Chinnery, P.F. Adult-Onset Spinocerebellar Ataxia Syndromes Due to MTATP6 Mutations. J. Neurol. Neurosurg. Psychiatry 2012, 83, 883–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, R.; Tozawa, T.; Kondo, H.; Kizaki, Z.; Kishita, Y.; Okazaki, Y.; Murayama, K.; Ohtake, A.; Chiyonobu, T. Early Infantile-Onset Leigh Syndrome Complicated with Infantile Spasms Associated with the m.9185 T > C Variant in the MT-ATP6 Gene: Expanding the Clinical Spectrum. Brain Dev. 2020, 42, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Wang, L.; Fei, G.; Dong, J.; Zhong, C.; Lu, J.; Jin, L. Acute Respiratory Failure Is the Initial Manifestation in the Adult-Onset A3243G TRNALeu MtDNA Mutation: A Case Report and the Literature Review. Front. Neurol. 2019, 10, 780. [Google Scholar] [CrossRef] [Green Version]
- Mahale, R.R.; Gautham, J.; Mailankody, P.; Padmanabha, H.; Mathuranath, P.S. Isolated Mitochondrial Myopathy Due to m.3243A > G Mutation in MT-TL1 Gene. Acta Neurol. Belg. 2022, 122, 1115–1116. [Google Scholar] [CrossRef]
- Esterhuizen, K.; Lindeque, J.Z.; Mason, S.; Van Der Westhuizen, F.H.; Rodenburg, R.J.; De Laat, P.; Smeitink, J.A.M.; Janssen, M.C.H.; Louw, R. One Mutation, Three Phenotypes: Novel Metabolic Insights on MELAS, MIDD and Myopathy Caused by the m.3243A > G Mutation. Metabolomics 2021, 17, 10. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Taylor, D.J.; Brown, D.T.; Manners, D.; Styles, P.; Lodi, R. Very Low Levels of the MtDNA A3243G Mutation Associated with Mitochondrial Dysfunction in Vivo. Ann. Neurol. 2000, 47, 381–384. [Google Scholar] [CrossRef]
- Leshinsky-Silver, E.; Lev, D.; Tzofi-Berman, Z.; Cohen, S.; Saada, A.; Yanoov-Sharav, M.; Gilad, E.; Lerman-Sagie, T. Fulminant Neurological Deterioration in a Neonate with Leigh Syndrome Due to a Maternally Transmitted Missense Mutation in the Mitochondrial ND3 Gene. Biochem. Biophys. Res. Commun. 2005, 334, 582–587. [Google Scholar] [CrossRef]
- Malfatti, E.; Bugiani, M.; Invernizzi, F.; De Souza, C.F.-M.; Farina, L.; Carrara, F.; Lamantea, E.; Antozzi, C.; Confalonieri, P.; Sanseverino, M.T.; et al. Novel Mutations of ND Genes in Complex I Deficiency Associated with Mitochondrial Encephalopathy. Brain 2007, 130, 1894–1904. [Google Scholar] [CrossRef]
- Newstead, S.M.; Scorza, C.A.; Fiorini, A.C.; Scorza, F.A.; Finsterer, J. Mitochondrial Small Fiber Neuropathy as a Novel Phenotypic Trait of Leigh-like Syndrome Due to the Variant m.10191T>C in MT-ND3. Clin. Sao Paulo Braz. 2023, 78, 100206. [Google Scholar] [CrossRef] [PubMed]
- Swalwell, H.; Kirby, D.M.; Blakely, E.L.; Mitchell, A.; Salemi, R.; Sugiana, C.; Compton, A.G.; Tucker, E.J.; Ke, B.-X.; Lamont, P.J.; et al. Respiratory Chain Complex I Deficiency Caused by Mitochondrial DNA Mutations. Eur. J. Hum. Genet. 2011, 19, 769–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newstead, S.M.; Finsterer, J. Leigh-Like Syndrome with a Novel, Complex Phenotype Due to m.10191T>C in Mt-ND3. Cureus 2022, 14, e28986. [Google Scholar] [CrossRef]
- Lax, N.Z.; Pienaar, I.S.; Reeve, A.K.; Hepplewhite, P.D.; Jaros, E.; Taylor, R.W.; Kalaria, R.N.; Turnbull, D.M. Microangiopathy in the Cerebellum of Patients with Mitochondrial DNA Disease. Brain 2012, 135, 1736–1750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, E.; Shimura, M.; Fushimi, T.; Tajika, M.; Ichimoto, K.; Matsunaga, A.; Tsuruoka, T.; Ishige, M.; Fuchigami, T.; Yamazaki, T.; et al. Clinical Validity of Biochemical and Molecular Analysis in Diagnosing Leigh Syndrome: A Study of 106 Japanese Patients. J. Inherit. Metab. Dis. 2017, 40, 685–693. [Google Scholar] [CrossRef] [Green Version]
- Ardissone, A.; Ferrera, G.; Lamperti, C.; Tiranti, V.; Ghezzi, D.; Moroni, I.; Lamantea, E. Phenotyping mitochondrial DNA-related Diseases in Childhood: A Cohort Study of 150 Patients. Eur. J. Neurol. 2023, 30, 2079–2091. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, E.M.; Siers, M.H.; Van Den Elzen, C.; Van Engelen, B.G.; Smeitink, J.A.M.; Rodenburg, R.J.; Hol, F.A. The Mitochondrial 13513G>A Mutation Is Most Frequent in Leigh Syndrome Combined with Reduced Complex I Activity, Optic Atrophy and/or Wolff–Parkinson–White. Eur. J. Hum. Genet. 2007, 15, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, A.; Falk, M.J. Mitochondrial DNA Deletion Syndromes; University of Washington: Seattle, WA, USA, 2019. [Google Scholar]
- Reynolds, E.; Byrne, M.; Ganetzky, R.; Parikh, S. Pediatric Single Large-Scale MtDNA Deletion Syndromes: The Power of Patient Reported Outcomes. Mol. Genet. Metab. 2021, 134, 301–308. [Google Scholar] [CrossRef]
- McShane, M.A.; Hardingt, A.E. Pearson Syndrome and Mitochondrial Encephalomyopathy in a Patient with a Deletion of MtDNA. Am. J. Hum. Genet. 1991, 48, 39. [Google Scholar] [PubMed]
- Rötig, A.; Bourgeron, T.; Chretien, D.; Rustin, P.; Munnich, A. Spectrum of Mitochondrial DNA Rearrangements in the Pearson Marrow-Pancreas Syndrome. Hum. Mol. Genet. 1995, 4, 1327–1330. [Google Scholar] [CrossRef]
- Incecik, F.; Herguner, O.; Besen, S.; Bozdoğan, S.; Mungan, N. Late-Onset Leigh Syndrome Due to NDUFV1 Mutation in a 10-Year-Old Boy Initially Presenting with Ataxia. J. Pediatr. Neurosci. 2018, 13, 205. [Google Scholar] [CrossRef]
- Fassone, E.; Rahman, S. Complex I Deficiency: Clinical Features, Biochemistry and Molecular Genetics. J. Med. Genet. 2012, 49, 578–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Men, L.; Feng, J.; Huang, W.; Xu, M.; Zhao, X.; Sun, R.; Xu, J.; Cao, L. Lip Cyanosis as the First Symptom of Leigh Syndrome Associated with Mitochondrial Complex I Deficiency Due to a Compound Heterozygous NDUFS1 Mutation: A Case Report. Med. (Baltim.) 2022, 101, e30303. [Google Scholar] [CrossRef]
- Koene, S.; Rodenburg, R.J.; Van Der Knaap, M.S.; Willemsen, M.A.A.P.; Sperl, W.; Laugel, V.; Ostergaard, E.; Tarnopolsky, M.; Martin, M.A.; Nesbitt, V.; et al. Natural Disease Course and Genotype-Phenotype Correlations in Complex I Deficiency Caused by Nuclear Gene Defects: What We Learned from 130 Cases. J. Inherit. Metab. Dis. 2012, 35, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Loeffen, J.L.C.M.; Smeitink, J.A.M.; Trijbels, J.M.F.; Janssen, A.J.M.; Triepels, R.H.; Sengers, R.C.A.; Van Den Heuvel, L.P. Isolated Complex I Deficiency in Children: Clinical, Biochemical and Genetic Aspects. Hum. Mutat. 2000, 15, 123–134. [Google Scholar] [CrossRef]
- Werner, K.G.E.; Morel, C.F.; Kirton, A.; Benseler, S.M.; Shoffner, J.M.; Addis, J.B.L.; Robinson, B.H.; Burrowes, D.M.; Blaser, S.I.; Epstein, L.G.; et al. Rolandic Mitochondrial Encephalomyelopathy and MT-ND3 Mutations. Pediatr. Neurol. 2009, 41, 27–33. [Google Scholar] [CrossRef]
- Pagniez-Mammeri, H.; Lombes, A.; Brivet, M.; Ogier-de Baulny, H.; Landrieu, P.; Legrand, A.; Slama, A. Rapid Screening for Nuclear Genes Mutations in Isolated Respiratory Chain Complex I Defects. Mol. Genet. Metab. 2009, 96, 196–200. [Google Scholar] [CrossRef]
- Cameron, J.M.; MacKay, N.; Feigenbaum, A.; Tarnopolsky, M.; Blaser, S.; Robinson, B.H.; Schulze, A. Exome Sequencing Identifies Complex I NDUFV2 Mutations as a Novel Cause of Leigh Syndrome. Eur. J. Paediatr. Neurol. 2015, 19, 525–532. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, L.; Ren, C.; Xu, M.; Li, S.; Ban, R.; Wu, Y.; Chen, L.; Sun, S.; Elstner, M.; et al. Whole Genome and Exome Sequencing Identify NDUFV2 Mutations as a New Cause of Progressive Cavitating Leukoencephalopathy. J. Med. Genet. 2022, 59, 351–357. [Google Scholar] [CrossRef]
- Shoubridge, E.A. Cytochrome c Oxidase Deficiency. Am. J. Med. Genet. 2001, 106, 46–52. [Google Scholar] [CrossRef]
- Oláhová, M.; Hardy, S.A.; Hall, J.; Yarham, J.W.; Haack, T.B.; Wilson, W.C.; Alston, C.L.; He, L.; Aznauryan, E.; Brown, R.M.; et al. LRPPRC Mutations Cause Early-Onset Multisystem Mitochondrial Disease Outside of the French-Canadian Population. Brain 2015, 138, 3503–3519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baertling, F.; Mayatepek, E.; Distelmaier, F. Hypertrichosis in Presymptomatic Mitochondrial Disease. J. Inherit. Metab. Dis. 2013, 36, 1081–1082. [Google Scholar] [CrossRef]
- Wedatilake, Y.; Brown, R.M.; McFarland, R.; Yaplito-Lee, J.; Morris, A.A.M.; Champion, M.; Jardine, P.E.; Clarke, A.; Thorburn, D.R.; Taylor, R.W.; et al. SURF1 Deficiency: A Multi-Centre Natural History Study. Orphanet J. Rare Dis. 2013, 8, 96. [Google Scholar] [CrossRef] [Green Version]
- Mahdieh, N.; Soveizi, M.; Tavasoli, A.R.; Rabbani, A.; Ashrafi, M.R.; Kohlschütter, A.; Rabbani, B. Genetic Testing of Leukodystrophies Unraveling Extensive Heterogeneity in a Large Cohort and Report of Five Common Diseases and 38 Novel Variants. Sci. Rep. 2021, 11, 3231. [Google Scholar] [CrossRef]
- Debray, F.-G.; Morin, C.; Janvier, A.; Villeneuve, J.; Maranda, B.; Laframboise, R.; Lacroix, J.; Decarie, J.-C.; Robitaille, Y.; Lambert, M.; et al. LRPPRC Mutations Cause a Phenotypically Distinct Form of Leigh Syndrome with Cytochrome c Oxidase Deficiency. J. Med. Genet. 2011, 48, 183–189. [Google Scholar] [CrossRef]
- Distelmaier, F.; Huppke, P.; Pieperhoff, P.; Amunts, K.; Schaper, J.; Morava, E.; Mayatepek, E.; Kohlhase, J.; Karenfort, M. Biotin-Responsive Basal Ganglia Disease: A Treatable Differential Diagnosis of Leigh Syndrome. In JIMD Reports-Case and Research Reports; Zschocke, J., Gibson, K.M., Brown, G., Morava, E., Peters, V., Eds.; JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2013; Volume 13, pp. 53–57. [Google Scholar] [CrossRef] [Green Version]
- Ortigoza-Escobar, J.D.; Molero-Luis, M.; Arias, A.; Oyarzabal, A.; Darín, N.; Serrano, M.; Garcia-Cazorla, A.; Tondo, M.; Hernández, M.; Garcia-Villoria, J.; et al. Free-Thiamine Is a Potential Biomarker of Thiamine Transporter-2 Deficiency: A Treatable Cause of Leigh Syndrome. Brain 2016, 139, 31–38. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Han, X.; Liu, Z.; Ma, Y.; Chen, G.; Zhang, H.; Sun, D.; Xu, R.; Liu, Y.; et al. Report of the Largest Chinese Cohort with SLC19A3 Gene Defect and Literature Review. Front. Genet. 2021, 12, 683255. [Google Scholar] [CrossRef] [PubMed]
- Tabarki, B.; Al-Hashem, A.; Alfadhel, M. Biotin-Thiamine-Responsive Basal Ganglia Disease; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Olgiati, S.; Skorvanek, M.; Quadri, M.; Minneboo, M.; Graafland, J.; Breedveld, G.J.; Bonte, R.; Ozgur, Z.; Van Den Hout, M.C.G.N.; Schoonderwoerd, K.; et al. Paroxysmal Exercise-Induced Dystonia within the Phenotypic Spectrum of ECHS1 Deficiency: ECHS1 Mutations, Dystonia, and PED. Mov. Disord. 2016, 31, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Ganetzky, R.D.; Bloom, K.; Ahrens-Nicklas, R.; Edmondson, A.; Deardorff, M.A.; Bennett, M.J.; Ficicioglu, C. ECHS1 Deficiency as a Cause of Severe Neonatal Lactic Acidosis. In JIMD Reports; Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; Volume 30, pp. 33–37. [Google Scholar] [CrossRef] [Green Version]
- Ferdinandusse, S.; Waterham, H.R.; Heales, S.J.; Brown, G.K.; Hargreaves, I.P.; Taanman, J.-W.; Gunny, R.; Abulhoul, L.; Wanders, R.J.; Clayton, P.T.; et al. HIBCH Mutations Can Cause Leigh-like Disease with Combined Deficiency of Multiple Mitochondrial Respiratory Chain Enzymes and Pyruvate Dehydrogenase. Orphanet J. Rare Dis. 2013, 8, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Çakar, N.; Görükmez, O. 3-Hydroxyisobutyryl-CoA Hydrolase (HIBCH) Deficiency Cases Diagnosed by Only HIBCH Gene Analysis and Novel Pathogenic Mutation. Ann. Indian Acad. Neurol. 2021, 24, 372. [Google Scholar] [CrossRef] [PubMed]
- Marti-Sanchez, L.; Baide-Mairena, H.; Marcé-Grau, A.; Pons, R.; Skouma, A.; López-Laso, E.; Sigatullina, M.; Rizzo, C.; Semeraro, M.; Martinelli, D.; et al. Delineating the Neurological Phenotype in Children with Defects in the ECHS1 or HIBCH Gene. J. Inherit. Metab. Dis. 2021, 44, 401–414. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Leigh Syndrome Spectrum (LSS) | |
|---|---|
| Clinical features * | Metabolic decompensation (elevated lactate levels in blood and/or cerebrospinal fluid) and/or characteristic symptoms during acute illness (episodic). Neurologic manifestations: hypotonia, spasticity, movement disorders (dystonia, chorea), cerebellar ataxia, and peripheral neuropathy. Other systems manifestations: hypertrophic cardiomyopathy, anemia, renal tubulopathy, liver involvement, ptosis, and muscle weakness. |
| Radiological features | Multiple symmetric bilateral lesions in the basal ganglia, thalamus, brain stem, dentate nuclei, and optic nerves **. |
| Gender | Age of Diagnosis | Gene | Mutation Data | Symptoms | ||||
|---|---|---|---|---|---|---|---|---|
| Variant | Heteroplasmy | Status (Mitomap) [22] | Reference | |||||
| P1 | F | 7 y | MT-ATP6 | m.8993T>G (p.Leu156Arg) | Muscle: 95% | P | Holt, 1990 [4] | Psychomotor regression, loss of contact, lethargy, generalized seizures, dystonia. |
| P2 | F | 2 y | MT-ATP6 | m.8993T>G (p.Leu156Arg) | Muscle: homoplasmic | P | Holt, 1990 [4] | Hypotonia, progression to apnea after febrile illness, dystonia. |
| P3 | F | 8 y | MT-ATP6 | m.8993T>G (p.Leu156Arg) | Blood: homoplasmic | P | Holt, 1990 [4] | Intellectual impairment, spastic paraparesis, hyperlactacidemia. |
| P4 | F | 9 y | MT-ATP6 | m.8993T>G (p.Leu156Arg) | Blood: 95% | P | Holt, 1990 [4] | Mitochondrial encephalopathy. |
| P5 | M | 1 y | MT-ATP6 | m.8993T>G (p.Leu156Arg) | Muscle: 95% | P | Holt, 1990 [4] | Hypotonia, myoclonic epilepsy, regression of milestones. |
| P6 | F | 29 y | MT-ATP6 | m.8993T>G (p.Leu156Arg) | Blood and muscle: >95% | P | Holt, 1990 [4] | LSS |
| P7 | F | 16 y | MT-ATP6 | m.8993T>G (p.Leu156Arg) | Muscle: 75% | P | Holt, 1990 [4] | LSS |
| P8 | M | 6 y | MT-ATP6 | m.8993T>G (p.Leu156Arg) | Muscle: 85% | P | Holt, 1990 [4] | LSS |
| P9 | M | 1 y | MT-ATP6 | m.8993T>G (p.Leu156Arg) | Blood and muscle: homoplasmic | P | Holt, 1990 [4] | LSS |
| P10 | F | 1 y | MT-ATP6 | m.8993T>G (p.Leu156Arg) | Muscle: 90% | P | Holt, 1990 [4] | Neurodevelopmental delay, hypotonia, epilepsy. |
| P11 | F | 19 y | MT-ATP6 | m.8993T>G (p.Leu156Arg) | Muscle: 95% | P | Holt, 1990 [4] | Deafness, peripheral neuropathy, ataxia. |
| P12 | F | 2 y | MT-ATP6 | m.8993T>G (p.Leu156Arg) | Muscle: 98% | P | Holt, 1990 [4] | Failure to thrive, eosinophilic esophagitis. |
| P13 | F | 23 y | MT-ATP6 | m.8993T>C (p.Leu156Pro) | Muscle: >95% | P | De Vries, 1993 [23] | LSS |
| P14 | M | 8 y | MT-ATP6 | m.8993T>C (p.Leu156Pro) | Muscle: >95% | P | De Vries, 1993 [23] | LSS |
| P15 | F | 10 y | MT-ATP6 | m.8993T>C (p.Leu156Pro) | Blood and muscle: >90% | P | De Vries, 1993 [23] | Numbness, ataxia. |
| P16 | M | 8 y | MT-ATP6 | m.8993T>C (p.Leu156Pro) | Muscle: 98% | P | De Vries, 1993 [23] | Mitochondrial disorder. |
| P17 | F | 1 y | MT-ATP6 | m.9185T>C (p.Leu220Pro) | Muscle: 70% | P | Moslemi, 2005 [24] | Neurodevelopmental delay, microcephaly and hypotonia. |
| P18 | M | 32 y | MT-ATP7 | m.9185T>C (p.Leu220Pro) | Blood: homoplasmic | P | Moslemi, 2005 [24] | LSS |
| P19 | M | 0 m.o. | MT-ATP6 | m.9176T>G (p.Leu217Arg) | Blood: homoplasmic | P | Thyagarajan, 1995 [25] | Still asymptomatic (studied to clarify newborn screening alterations). |
| P20 | M | 10 y | MT-TL1 | m.3243A>G | Muscle: 73% | P | Poulton, 1988 [26] | Epilepsy, learning disability, ataxia, hyperlactacidemia. |
| P21 | F | 4 y | MT-ND3 | m.10191T>C (p.Ser45Pro) | Blood: 80%, muscle: 90% | P | Taylor, 2001 [6] | Hypotonia, neurodevelopmental delay, recurrent respiratory infections in first months of life, epilepsy. |
| P22 | M | 7 y | MT-ND3 | m.10197G>A (p.Ala47Tre) | Muscle: 100% | P | Kirby, 2004 [27] | Epilepsy, failure to thrive, recurrent illness, hypotonia, high serum lactate levels. |
| P23 | M | 11 y | MT-ND5 | m.13094T>C (p.Val253Ala) | Blood: 100% | P | Valente, 2009 [28] | Severe hypotonia. |
| P24 | F | 8 m.o. | MT-ND5 | m.13513G>A (p.Asp393Asn) | Blood: 70% | P | Chol, 2003 [29] | Neurodevelopmental delay, cardiomyopathy, hypotonia. |
| P25 | M | 7 y | MT-ND5 | m.13513G>A (p.Asp393Asn) | Blood: 58%, muscle: 88% | P | Chol, 2003 [29] | Ataxia and apnea. |
| P26 | M | 12 y | MT-ND1 | m.4142G>T (p.Arg279Leu) | Muscle: 80% | R | Pereira, 2019 [30] | Spastic deambulation with cerebellar signs, hyperreflexia, clonus, dysarthria, dystonia, cognitive impairment. |
| P27 | M | 13 y | MT-CO1 | m.6547T>C (p.Leu215Pro) | Muscle: 50% | R | Pereira, 2019 [30] | Hypotonia, hyperlactacidemia. |
| P28 | M | 7 y | MT-ATP6 to MT-ND5 | 4977bp-del (m.8470-m.13447) | Muscle: 61% | P | Vilarinho, 1997 [31] | Epilepsy, short stature, cerebellar ataxia, tremor. |
| Patient | Gender | Age of Diagnosis | Gene | Mutation Data | Symptoms | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allele 1 | Reference | ClinVar | ClinVar Phenotype | Allele 2 | Reference | ClinVar | ClinVar Phenotype | |||||
| P29 | M | 36 y | NDUFS8 | c.196C>T (p. Arg66*) | Nogueira, 2019 [22] | ND | ND | c.287G>A (p.Arg96 His) | Nogueira, 2019 [22] | ND | ND | Short stature, Leigh syndrome compatible neuroimaging. |
| P30 | M | 2 y | NDUFS1 | c.470A>T (p.Lys157 Met) | ND | ND | ND | c.1798G>C (p.Glu600 Gln) | ND | ND | ND | Axial hypotonia, failure to thrive, neurodevelopmental delay, nystagmus. |
| P31 | F | 2 y | NDUFV2 | c.120+5_ 120+8 delGT | Bénit, 2003 [36] | P | Mitochondrial Complex I deficiency | c.120+5_ 120+8 delGT | Bénit, 2003 [36] | P | Mitochondrial Complex I deficiency | Severe eczema, epilepsy, axial hypotonia, neurodevelopmental delay, suspicion of Leigh syndrome. |
| P32 | M | prenatal | c.120+5_ 120+8 delGT | Bénit, 2003 [36] | P | Mitochondrial Complex I deficiency | c.120+5_ 120+8 delGT | Bénit, 2003 [36] | P | Mitochondrial Complex I deficiency | Prenatal screening, P31 sibling. | |
| P33 | F | 3 y | SURF1 | c.19_35dup17* (p.Ala13 Cysfs*65) | Tiranti, 1998 [37] | P | LSS | c.845_846 del (p:Ser282 Cysfs*9) | Tiranti, 1998 [37] | P/LP | LSS | Ataxia, hypotonia, tremors, Tetralogy of Fallot. |
| P34 | F | 1 y | c.19_35dup17* (p.Ala13 Cysfs*65) | Tiranti, 1998 [37] | P | LSS | c.845_846 del (p:Ser282 Cysfs*9) | Tiranti, 1998 [37] | P/LP | LSS | Neurodevelopmental delay, hypotonia, bradycardia. | |
| P35 | M | 15 y | c.19_35dup17* (p.Ala13 Cysfs*65) | Tiranti, 1998 [37] | P | LSS | c.19_35dup17* (p.Ala13 Cysfs*65) | Tiranti, 1998 [37] | P | LSS | Visual hallucinations. | |
| P36 | F | 4 y | LRPPRC | c.74G>A (p.Arg25 His) | ND | VUS | Congenit lactic acidosis | c.74G>A (p. Arg25 His) | ND | VUS | Congenit lactic acidosis | Encephalopathy, brain atrophy. |
| P37 | F | 1y | SLC19A3 | c.74dupT (p.Ser26 Leufs*19) | Debs, 2010 [33] | P | BBGD | c.74dupT (p.Ser26 Leufs*19) | Debs, 2010 [33] | P | BBGD | Neonatal epileptic encephalopathy responsive to biotin, thiamine, Coenzyme Q10 and riboflavin. |
| P38 | F | 5y | c.980-14A>G | Debs, 2010 [33] | LP | BBGD | c.177G>A (p.Trp59*) | Debs, 2010 [33] | ND | ND | Episodic ataxia responsive to biotin, thiamine, Coenzyme Q10 and riboflavin. | |
| P39 | M | 3 m.o. | HIBCH | c.488G>T (p.Cys 163Phe) | Wojcik, 2019 [38] | LP | HIBCH deficiency | c.488G>T (p.Cys 163Phe) | Wojcik, 2019 [38] | LP | HIBCH deficiency | Stop of progression of the neurodevelopment and feeding difficulties. |
| P40 | F | 6 y | c.129dupA (p.Gly44 Argfs*20) | Peters, 2015 [35] | LP | HIBCH deficiency | c.910C>T (p.Pro304 Ser) | ND | ND | ND | Axial hypotonia, psychomotor regression after intercurrent illness, ataxia. | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baldo, M.S.; Nogueira, C.; Pereira, C.; Janeiro, P.; Ferreira, S.; Lourenço, C.M.; Bandeira, A.; Martins, E.; Magalhães, M.; Rodrigues, E.; et al. Leigh Syndrome Spectrum: A Portuguese Population Cohort in an Evolutionary Genetic Era. Genes 2023, 14, 1536. https://doi.org/10.3390/genes14081536
Baldo MS, Nogueira C, Pereira C, Janeiro P, Ferreira S, Lourenço CM, Bandeira A, Martins E, Magalhães M, Rodrigues E, et al. Leigh Syndrome Spectrum: A Portuguese Population Cohort in an Evolutionary Genetic Era. Genes. 2023; 14(8):1536. https://doi.org/10.3390/genes14081536
Chicago/Turabian StyleBaldo, Manuela Schubert, Célia Nogueira, Cristina Pereira, Patrícia Janeiro, Sara Ferreira, Charles M. Lourenço, Anabela Bandeira, Esmeralda Martins, Marina Magalhães, Esmeralda Rodrigues, and et al. 2023. "Leigh Syndrome Spectrum: A Portuguese Population Cohort in an Evolutionary Genetic Era" Genes 14, no. 8: 1536. https://doi.org/10.3390/genes14081536
APA StyleBaldo, M. S., Nogueira, C., Pereira, C., Janeiro, P., Ferreira, S., Lourenço, C. M., Bandeira, A., Martins, E., Magalhães, M., Rodrigues, E., Santos, H., Ferreira, A. C., & Vilarinho, L. (2023). Leigh Syndrome Spectrum: A Portuguese Population Cohort in an Evolutionary Genetic Era. Genes, 14(8), 1536. https://doi.org/10.3390/genes14081536