
Refinement of Leishmania donovani Genome Annotations in the Light of Ribosome-Protected mRNAs Fragments (Ribo-Seq Data)

 ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ribosome Footprint Data
2.2. Processing and Mapping of Reads
2.3. Preparation of a Database for All Possible ORFs
2.4. Identification of Ribosome-Protected uORFs
2.5. BLAST Searches Using the TriTryDB Repository
2.6. Data Availability
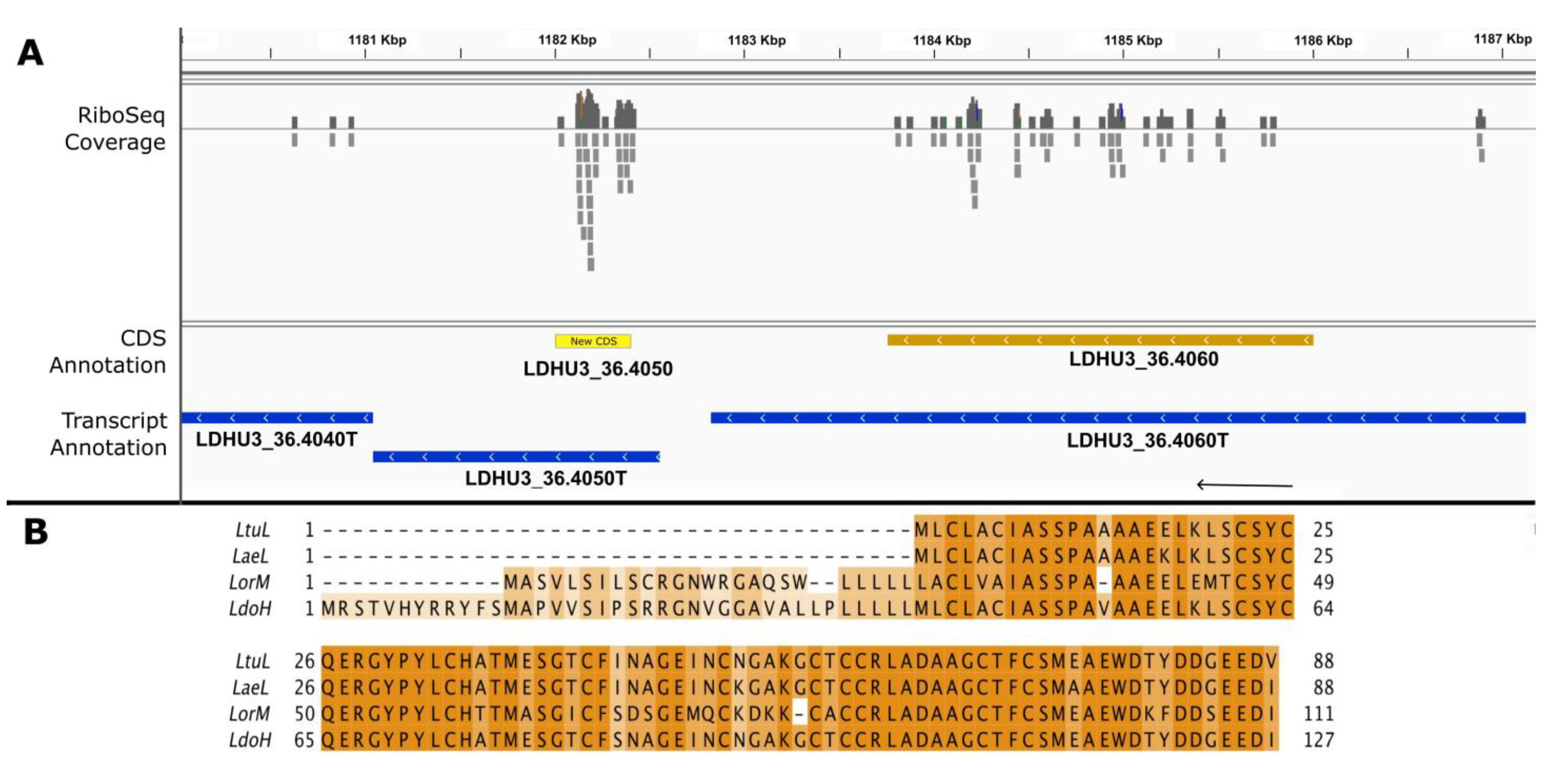
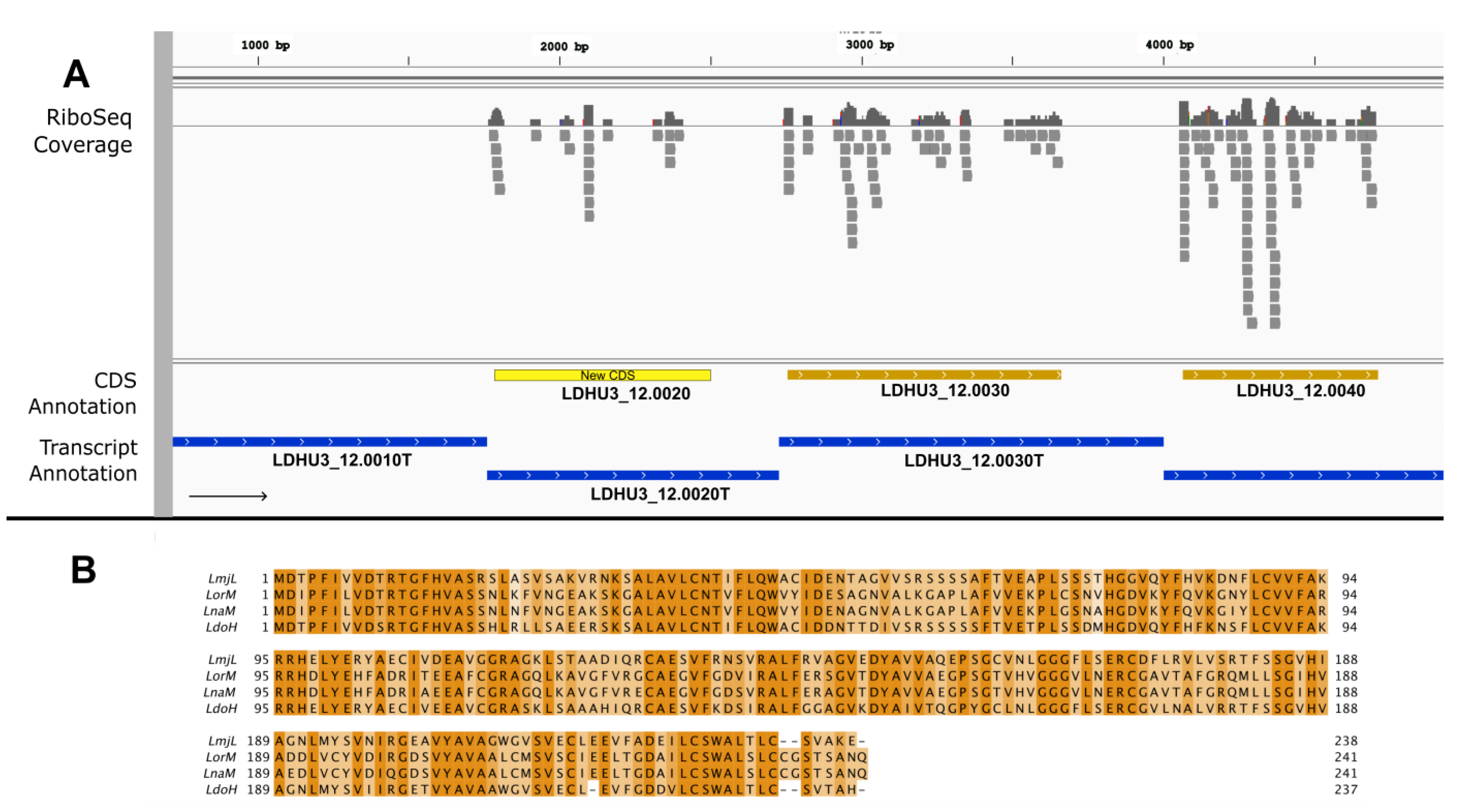
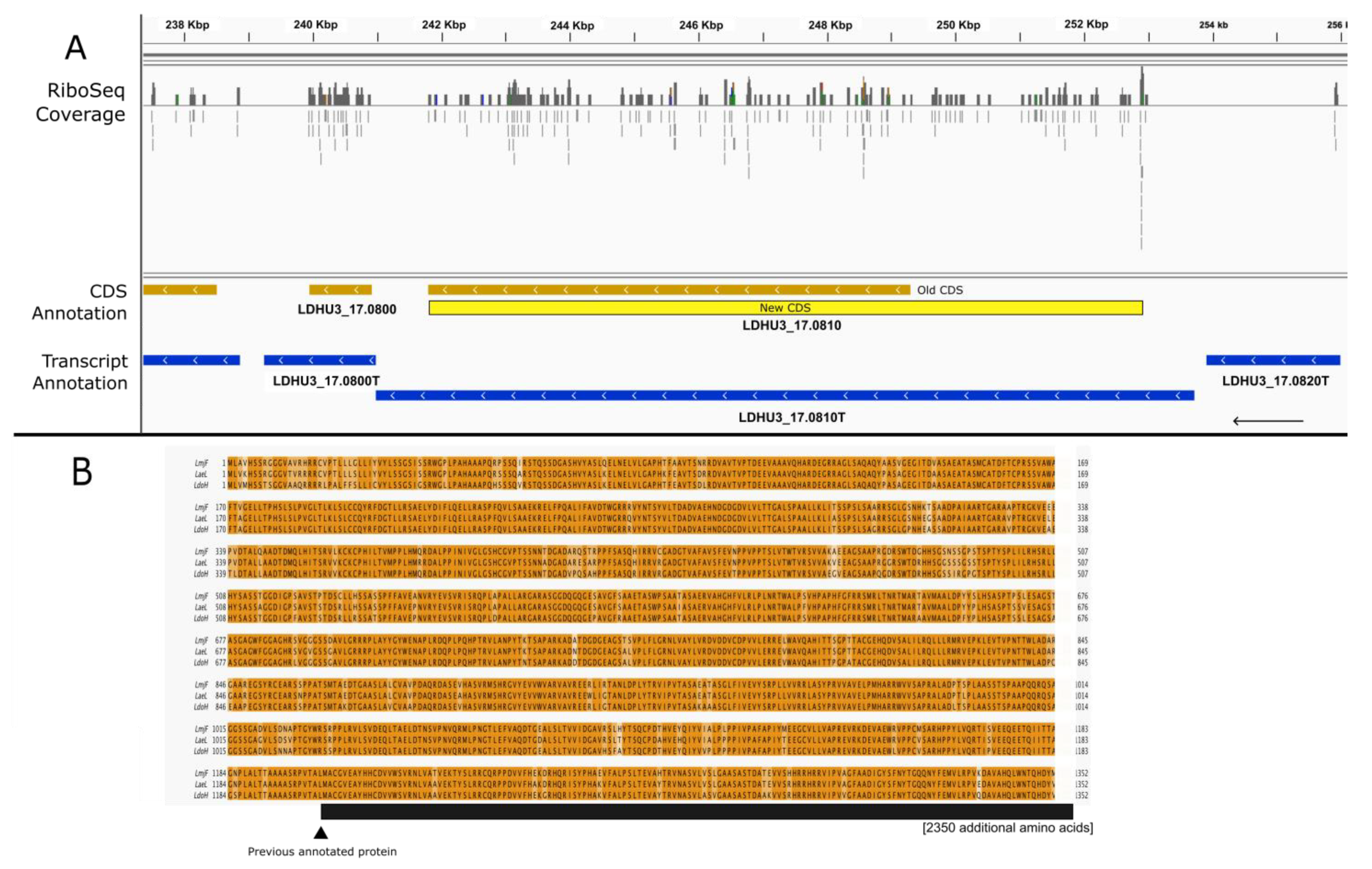
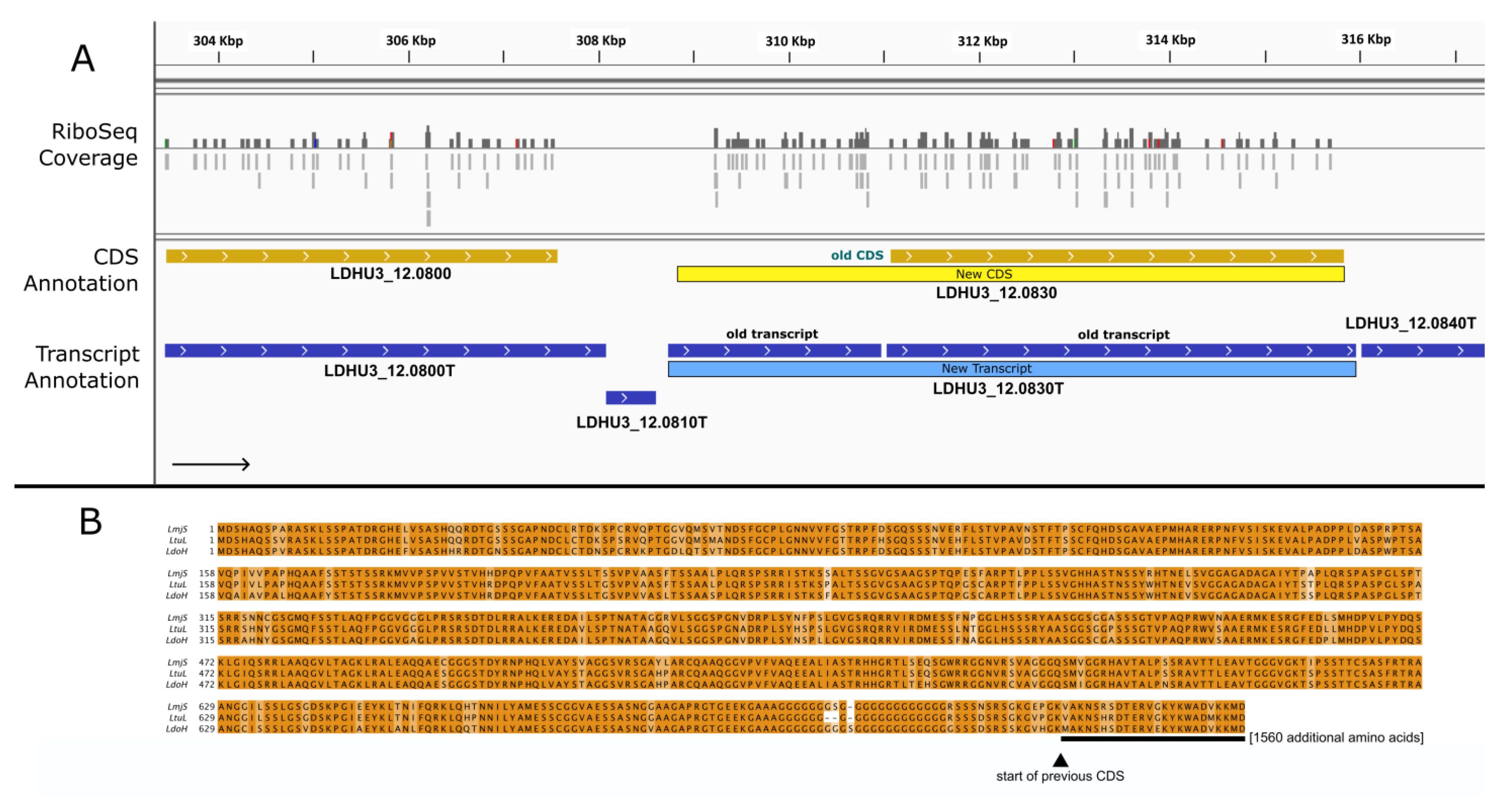
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alvar, J.; Velez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Postigo, J.A.; Saurabh, J.; Mikhailov, A.; Maia-Elkhoury, A.N.; Valadas, S.; Warusavithana, S.; Osman, M.; Lin, Z.; Beshah, A.; Yajima, A.; et al. Global leishmaniasis surveillance: 2019–2020, a baseline for the 2030 roadmap. Wkly. Epidemiol. Rec. 2021, 35, 401–419. [Google Scholar]
- McCall, L.I.; Zhang, W.W.; Matlashewski, G. Determinants for the development of visceral leishmaniasis disease. PLoS Pathog. 2013, 9, e1003053. [Google Scholar] [CrossRef] [PubMed]
- Ivens, A.C.; Peacock, C.S.; Worthey, E.A.; Murphy, L.; Aggarwal, G.; Berriman, M.; Sisk, E.; Rajandream, M.A.; Adlem, E.; Aert, R.; et al. The Genome of the Kinetoplastid Parasite, Leishmania major. Science 2005, 309, 436–442. [Google Scholar] [CrossRef]
- Camacho, E.; González-de la Fuente, S.; Solana, J.C.; Rastrojo, A.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Gene annotation and transcriptome delineation on a de novo genome assembly for the reference Leishmania major Friedlin strain. Genes 2021, 12, 1359. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundram, A.; Starns, D.; Böhme, U.; Amos, B.; Wilkinson, P.A.; Harb, O.S.; Warrenfeltz, S.; Kissinger, J.C.; McDowell, M.A.; Roos, D.S.; et al. TriTrypDB: An integrated functional genomics resource for kinetoplastida. PLoS Negl. Trop. Dis. 2023, 17, e0011058. [Google Scholar] [CrossRef]
- Stechmann, A.; Cavalier-Smith, T. Phylogenetic analysis of eukaryotes using heat-shock protein Hsp90. J. Mol. Evol. 2003, 57, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Steinbiss, S.; Silva-Franco, F.; Brunk, B.; Foth, B.; Hertz-Fowler, C.; Berriman, M.; Otto, T.D. Companion: A web server for annotation and analysis of parasite genomes. Nucleic Acids Res. 2016, 44, W29–W34. [Google Scholar] [CrossRef]
- Rastrojo, A.; Carrasco-Ramiro, F.; Martín, D.; Crespillo, A.; Reguera, R.M.; Aguado, B.; Requena, J.M. The transcriptome of Leishmania major in the axenic promastigote stage: Transcript annotation and relative expression levels by RNA-seq. BMC Genom. 2013, 14, 223. [Google Scholar] [CrossRef]
- Fiebig, M.; Kelly, S.; Gluenz, E. Comparative Life Cycle Transcriptomics Revises Leishmania mexicana Genome Annotation and Links a Chromosome Duplication with Parasitism of Vertebrates. PLoS Pathog. 2015, 11, e1005186. [Google Scholar] [CrossRef]
- Camacho, E.; González-de la Fuente, S.; Rastrojo, A.; Peiró-Pastor, R.; Solana, J.C.; Tabera, L.; Gamarro, F.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Complete assembly of the Leishmania donovani (HU3 strain) genome and transcriptome annotation. Sci. Rep. 2019, 9, 6127. [Google Scholar] [CrossRef] [PubMed]
- Sanchiz, Á.; Morato, E.; Rastrojo, A.; Camacho, E.; González-de la Fuente, S.; Marina, A.; Aguado, B.; Requena, J.M. The Experimental Proteome of Leishmania infantum Promastigote and Its Usefulness for Improving Gene Annotations. Genes 2020, 11, 1036. [Google Scholar] [CrossRef] [PubMed]
- Ingolia, N.T.; Brar, G.A.; Rouskin, S.; McGeachy, A.M.; Weissman, J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012, 7, 1534–1550. [Google Scholar] [CrossRef] [PubMed]
- Smircich, P.; Eastman, G.; Bispo, S.; Duhagon, M.A.; Guerra-Slompo, E.P.; Garat, B.; Goldenberg, S.; Munroe, D.J.; Dallagiovanna, B.; Holetz, F.; et al. Ribosome profiling reveals translation control as a key mechanism generating differential gene expression in Trypanosoma cruzi. BMC Genom. 2015, 16, 443. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.; Ramasamy, G.; Vasconcelos, E.J.; Jensen, B.C.; Myler, P.J. Advancing Trypanosoma brucei genome annotation through ribosome profiling and spliced leader mapping. Mol. Biochem. Parasitol. 2015, 202, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.C.; Ramasamy, G.; Vasconcelos, E.J.; Ingolia, N.T.; Myler, P.J.; Parsons, M. Extensive stage-regulation of translation revealed by ribosome profiling of Trypanosoma brucei. BMC Genom. 2014, 15, 911. [Google Scholar] [CrossRef]
- Vasquez, J.J.; Hon, C.C.; Vanselow, J.T.; Schlosser, A.; Siegel, T.N. Comparative ribosome profiling reveals extensive translational complexity in different Trypanosoma brucei life cycle stages. Nucleic Acids Res. 2014, 42, 3623–3637. [Google Scholar] [CrossRef]
- Bifeld, E.; Lorenzen, S.; Bartsch, K.; Vasquez, J.J.; Siegel, T.N.; Clos, J. Ribosome Profiling Reveals HSP90 Inhibitor Effects on Stage-Specific Protein Synthesis in Leishmania donovani. mSystems 2018, 3, e00214–e00218. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Anders, S.; McCarthy, D.J.; Chen, Y.; Okoniewski, M.; Smyth, G.K.; Huber, W.; Robinson, M.D. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 2013, 8, 1765–1786. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Amos, B.; Aurrecoechea, C.; Barba, M.; Barreto, A.; Basenko, E.Y.; Bażant, W.; Belnap, R.; Blevins, A.S.; Böhme, U.; Brestelli, J.; et al. VEuPathDB: The eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 2022, 50, D898–D911. [Google Scholar] [CrossRef]
- Gonzalez-de la Fuente, S.; Peiro-Pastor, R.; Rastrojo, A.; Moreno, J.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Resequencing of the Leishmania infantum (strain JPCM5) genome and de novo assembly into 36 contigs. Sci. Rep. 2017, 7, 18050. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Br. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Young, S.K.; Wek, R.C. Upstream Open Reading Frames Differentially Regulate Gene-specific Translation in the Integrated Stress Response. J. Biol. Chem. 2016, 291, 16927–16935. [Google Scholar] [CrossRef] [PubMed]
- Carter, M.; Gomez, S.; Gritz, S.; Larson, S.; Silva-Herzog, E.; Kim, H.-S.; Schulz, D.; Hovel-Miner, G. A Trypanosoma brucei ORFeome-Based Gain-of-Function Library Identifies Genes That Promote Survival during Melarsoprol Treatment. mSphere 2020, 5, e00769. [Google Scholar] [CrossRef]
- Gebauer, F.; Hentze, M.W. Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 2004, 5, 827–835. [Google Scholar] [CrossRef]
- Couso, J.P.; Patraquim, P. Classification and function of small open reading frames. Nat. Rev. Mol. Cell Biol. 2017, 18, 575–589. [Google Scholar] [CrossRef]
- Radío, S.; Garat, B.; Sotelo-Silveira, J.; Smircich, P. Upstream ORFs Influence Translation Efficiency in the Parasite Trypanosoma cruzi. Front. Genet. 2020, 11, 166. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Salvador, A.; González-de la Fuente, S.; Aguado, B.; Yates, P.A.; Requena, J.M. Refinement of Leishmania donovani Genome Annotations in the Light of Ribosome-Protected mRNAs Fragments (Ribo-Seq Data). Genes 2023, 14, 1637. https://doi.org/10.3390/genes14081637
Sánchez-Salvador A, González-de la Fuente S, Aguado B, Yates PA, Requena JM. Refinement of Leishmania donovani Genome Annotations in the Light of Ribosome-Protected mRNAs Fragments (Ribo-Seq Data). Genes. 2023; 14(8):1637. https://doi.org/10.3390/genes14081637
Chicago/Turabian StyleSánchez-Salvador, Alejandro, Sandra González-de la Fuente, Begoña Aguado, Phillip A. Yates, and Jose M. Requena. 2023. "Refinement of Leishmania donovani Genome Annotations in the Light of Ribosome-Protected mRNAs Fragments (Ribo-Seq Data)" Genes 14, no. 8: 1637. https://doi.org/10.3390/genes14081637
APA StyleSánchez-Salvador, A., González-de la Fuente, S., Aguado, B., Yates, P. A., & Requena, J. M. (2023). Refinement of Leishmania donovani Genome Annotations in the Light of Ribosome-Protected mRNAs Fragments (Ribo-Seq Data). Genes, 14(8), 1637. https://doi.org/10.3390/genes14081637