
Transcriptomic Analysis of Tobacco Plants in Response to Whitefly Infection

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plants and Whitefly
2.2. Plant Treatments
2.3. RNA Isolation and Illumina Sequencing
2.4. DEGs Expression Analysis
2.5. Quantitative Real-Time PCR (qRT-PCR)
3. Results
3.1. Transcriptomic Profile of Tobacco Plants Infested with Whiteflies
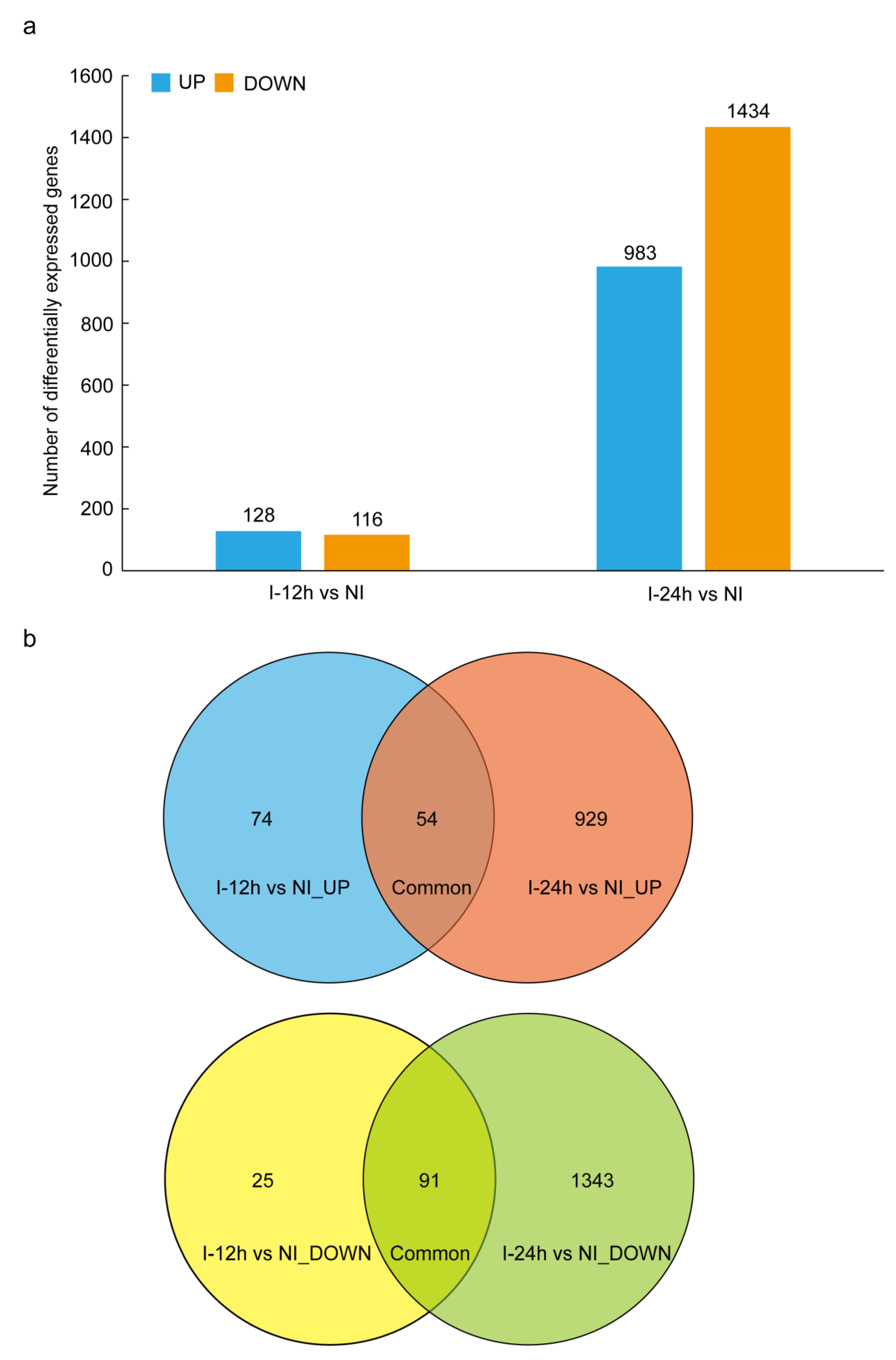
3.2. Differently Expressed Genes (DEGs) between Whitefly-Infested and Non-Infested Tobacco
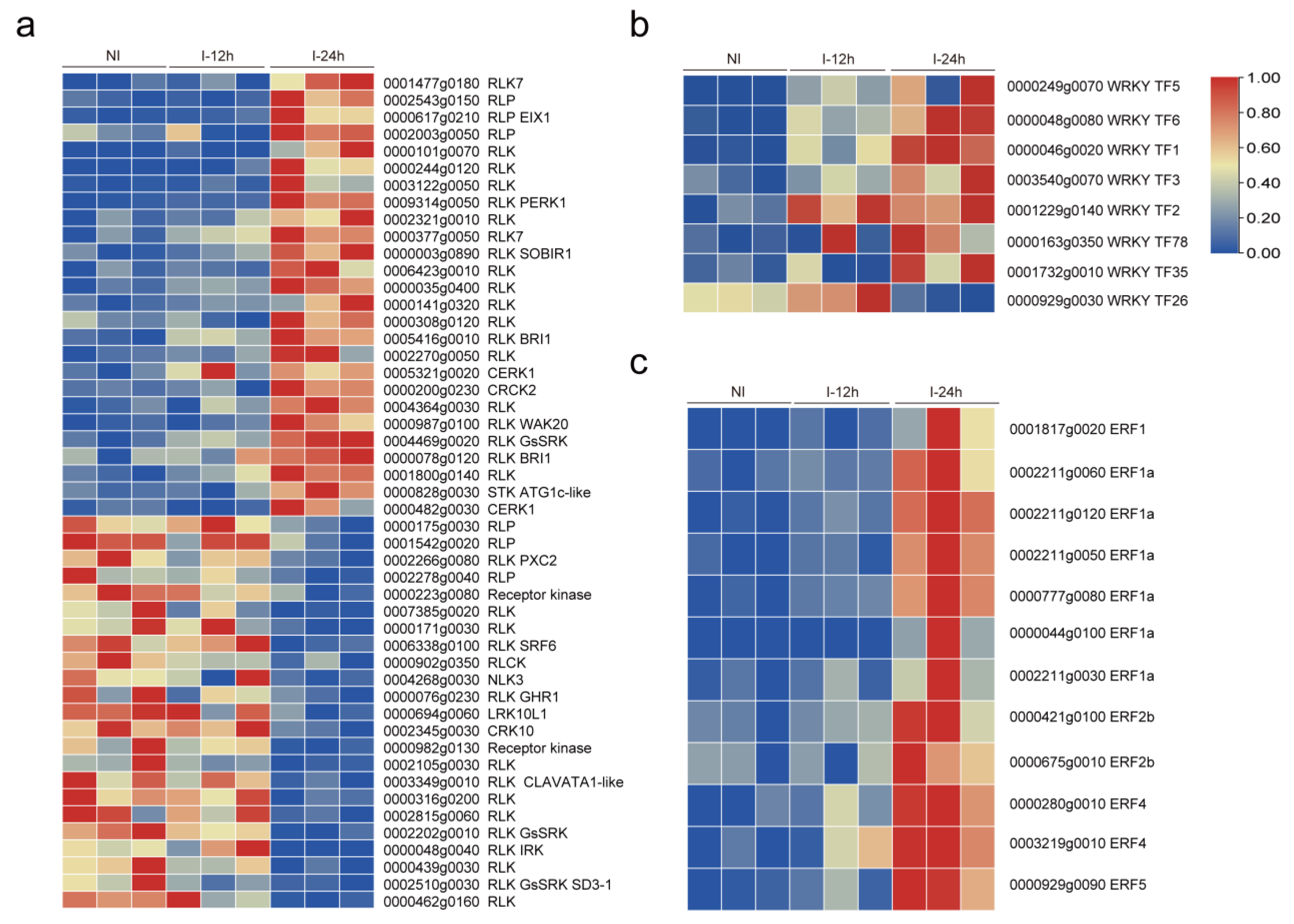
3.3. Plant Signal Networks Response to Whitefly Attacks
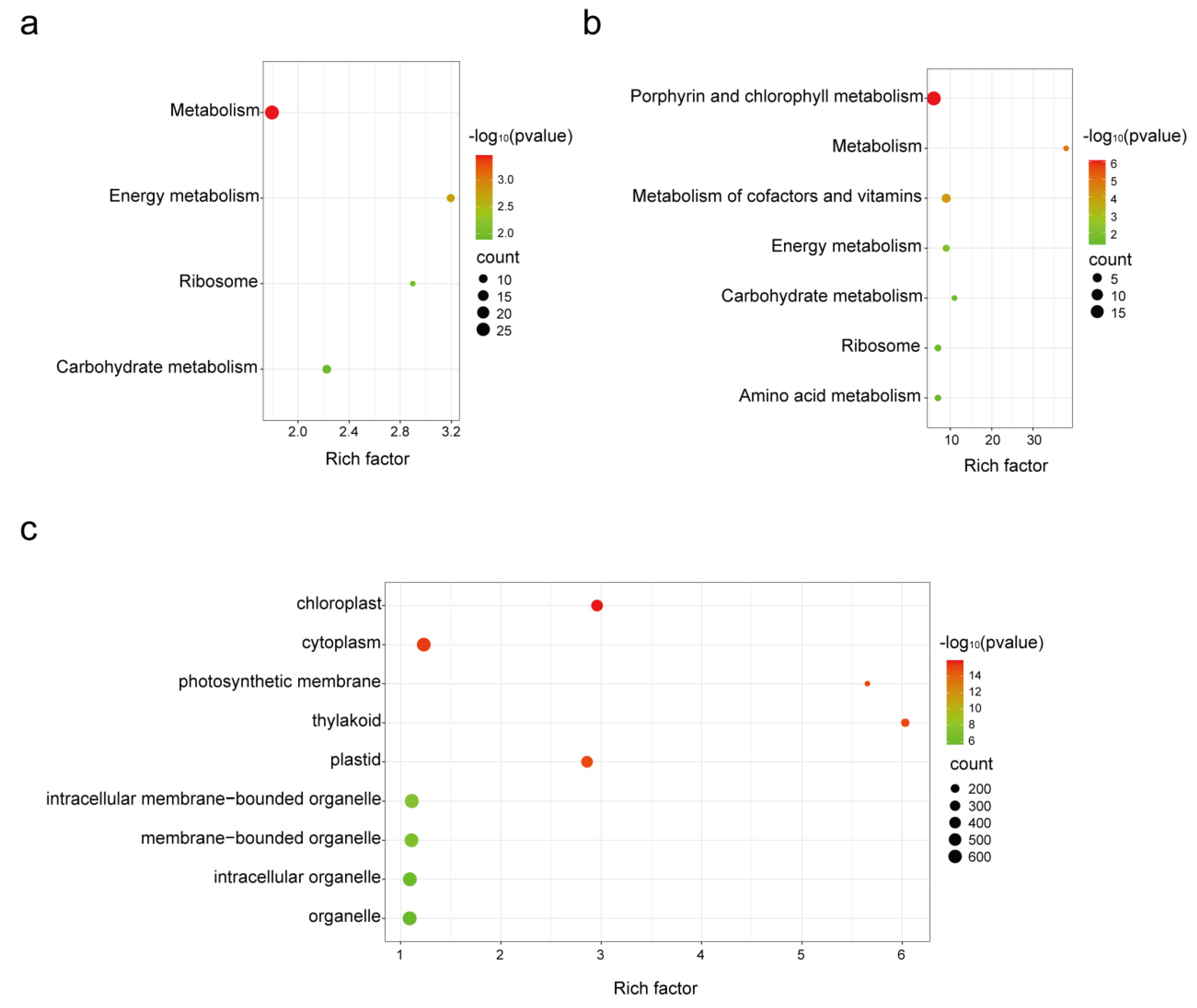
3.4. Metabolic Processes Affected by Whitefly Attack
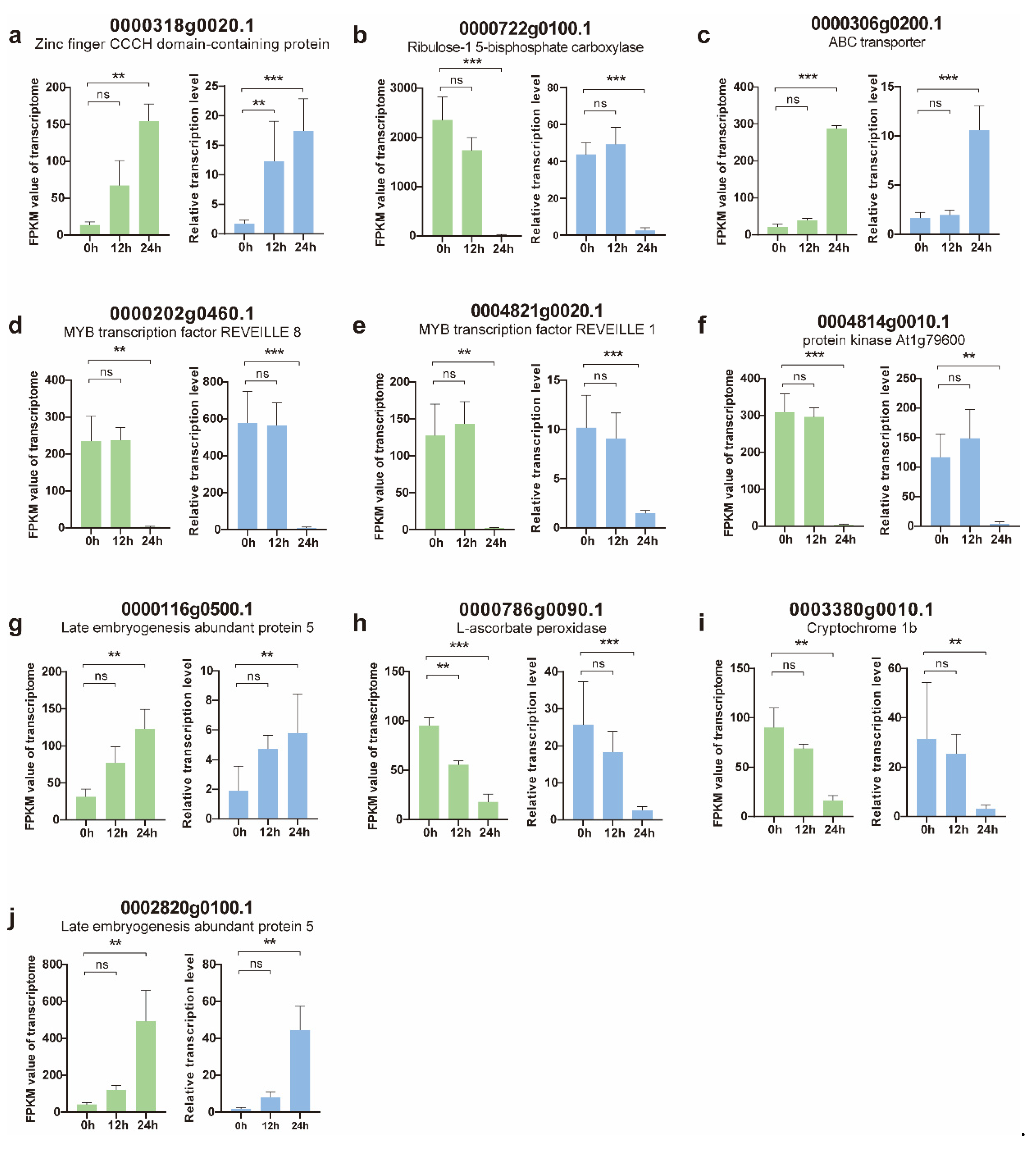
3.5. Validation of DEGs Using Quantitative Real-Time PCR (qRT-PCR)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Furstenberg-Hagg, J.; Zagrobelny, M.; Bak, S. Plant defense against insect herbivores. Int. J. Mol. Sci. 2013, 14, 10242–10297. [Google Scholar] [CrossRef] [PubMed]
- War, A.R.; Paulraj, M.G.; Ahmad, T.; Buhroo, A.A.; Hussain, B.; Ignacimuthu, S.; Sharma, H.C. Mechanisms of plant defense against insect herbivores. Plant Signal. Behav. 2012, 7, 1306–1320. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.S. Inducible direct plant defense against insect herbivores: A review. Insect Sci. 2008, 15, 101–114. [Google Scholar] [CrossRef]
- War, A.R.; Sharma, H.C.; Paulraj, M.G.; War, M.Y.; Ignacimuthu, S. Herbivore induced plant volatiles: Their role in plant defense for pest management. Plant Signal. Behav. 2011, 6, 1973–1978. [Google Scholar] [CrossRef]
- Arimura, G.; Ozawa, R.; Maffei, M.E. Recent advances in plant early signaling in response to herbivory. Int. J. Mol. Sci. 2011, 12, 3723–3739. [Google Scholar] [CrossRef]
- Wu, J.Q.; Baldwin, I.T. New insights into plant responses to the attack from insect herbivores. Annu. Rev. Genet. 2010, 44, 1–24. [Google Scholar] [CrossRef]
- Verma, V.; Ravindran, P.; Kumar, P.P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 2016, 16, 86. [Google Scholar] [CrossRef]
- Zipfel, C.; Felix, G. Plants and animals: A different taste for microbes? Curr. Opin. Plant Biol. 2005, 8, 353–360. [Google Scholar] [CrossRef]
- Nurnberger, T.; Brunner, F.; Kemmerling, B.; Piater, L. Innate immunity in plants and animals: Striking similarities and obvious differences. Immunol. Rev. 2004, 198, 249–266. [Google Scholar] [CrossRef]
- de Wit, P.J. How plants recognize pathogens and defend themselves. Cell. Mol. Life Sci. 2007, 64, 2726–2732. [Google Scholar] [CrossRef]
- Zipfel, C. Plant pattern-recognition receptors. Trends Immunol. 2014, 35, 345–351. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhou, J.; Shan, L.; Meng, X. Plant cell surface receptor-mediated signaling—A common theme amid diversity. J. Cell Sci. 2018, 131, jcs209353. [Google Scholar] [CrossRef] [PubMed]
- Dombrecht, B.; Xue, G.P.; Sprague, S.J.; Kirkegaard, J.A.; Ross, J.J.; Reid, J.B.; Fitt, G.P.; Sewelam, N.; Schenk, P.M.; Manners, J.M.; et al. MYC2 differentially modulates diverse jasmonate-dependent functions in Arabidopsis. Plant Cell 2007, 19, 2225–2245. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Ye, M.; Li, R.; Lou, Y. OsWRKY53, a versatile switch in regulating herbivore-induced defense responses in rice. Plant Signal. Behav. 2016, 11, e1169357. [Google Scholar] [CrossRef]
- Oliveira, M.; Henneberry, T.; Anderson, P. History, current status, and collaborative research projects for Bemisia tabaci. Crop Prot. 2001, 20, 709–723. [Google Scholar] [CrossRef]
- Navas-Castillo, J.; Fiallo-Olive, E.; Sanchez-Campos, S. Emerging virus diseases transmitted by whiteflies. Annu. Rev. Phytopathol. 2011, 49, 219–248. [Google Scholar] [CrossRef]
- Cock, M.J.W. Bemisia tabaci—A Literature Survey on the Cotton Whitefly with an Annotated Bibliography; C.A.B International Institute of Biological Control: Ascot, UK, 1986. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Andrews, S. FastQC a Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Babraham, UK, 2014. [Google Scholar]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Nomura, H.; Komori, T.; Uemura, S.; Kanda, Y.; Shimotani, K.; Nakai, K.; Furuichi, T.; Takebayashi, K.; Sugimoto, T.; Sano, S.; et al. Chloroplast-mediated activation of plant immune signaling in Arabidopsis. Nat. Commun. 2012, 3, 926. [Google Scholar] [CrossRef] [PubMed]
- Nomura, H.; Shiina, T. Calcium signaling in plant endosymbiotic organelles: Mechanism and role in physiology. Mol. Plant 2014, 7, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Muthamilarasan, M.; Prasad, M. Plant innate immunity: An updated insight into defense mechanism. J. Biosci. 2013, 38, 433–449. [Google Scholar] [CrossRef]
- Tang, D.Z.; Wang, G.X.; Zhou, J.M. Receptor kinases in plant-pathogen interactions: More than pattern recognition. Plant Cell 2017, 29, 618–637. [Google Scholar] [CrossRef]
- Reymond, P. Receptor kinases in plant responses to herbivory. Curr. Opin. Biotechnol. 2021, 70, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.H.; Ngou, B.P.M.; Ding, P.T.; Xiu-Fan, X. PTI-ETI crosstalk: An integrative view of plant immunity. Curr. Opin. Plant Biol. 2021, 62, 102030. [Google Scholar] [CrossRef]
- Devendrakumar, K.T.; Li, X.; Zhang, Y.L. MAP kinase signalling: Interplays between plant PAMP- and effector-triggered immunity. Cell. Mol. Life Sci. 2018, 75, 2981–2989. [Google Scholar] [CrossRef]
- Rasmussen, S.B.; Reinert, L.S.; Paludan, S.R. Innate recognition of intracellular pathogens: Detection and activation of the first line of defense. Apmis 2009, 117, 323–337. [Google Scholar] [CrossRef]
- Peng, Y.J.; van Wersch, R.; Zhang, Y.L. Convergent and divergent signaling in pamp-triggered immunity and effector-triggered immunity. Mol. Plant Microbe Interact. 2018, 31, 403–409. [Google Scholar] [CrossRef]
- Tsuda, K.; Katagiri, F. Comparing signaling mechanisms engaged in pattern-triggered and effector-triggered immunity. Curr. Opin. Plant Biol. 2010, 13, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Phukan, U.J.; Jeena, G.S.; Shukla, R.K. WRKY transcription factors: Molecular regulation and stress responses in plants. Front. Plant Sci. 2016, 7, 760. [Google Scholar] [CrossRef] [PubMed]
- Kundu, P.; Vadassery, J. Role of WRKY transcription factors in plant defense against lepidopteran insect herbivores: An overview. J. Plant Biochem. Biotechnol. 2021, 30, 698–707. [Google Scholar] [CrossRef]
- Kamphuis, L.G.; Zulak, K.; Gao, L.L.; Anderson, J.; Singh, K.B. Plant-aphid interactions with a focus on legumes. Funct. Plant Biol. 2013, 40, 1271–1284. [Google Scholar] [CrossRef]
- Li, J.; Zhu, L.; Hull, J.J.; Liang, S.; Daniell, H.; Jin, S.; Zhang, X. Transcriptome analysis reveals a comprehensive insect resistance response mechanism in cotton to infestation by the phloem feeding insect Bemisia tabaci (whitefly). Plant Biotechnol. J. 2016, 14, 1956–1975. [Google Scholar] [CrossRef]
- Wang, H.; Hao, J.; Chen, X.; Hao, Z.; Wang, X.; Lou, Y.; Peng, Y.; Guo, Z. Overexpression of rice WRKY89 enhances ultraviolet B tolerance and disease resistance in rice plants. Plant Mol. Biol. 2007, 65, 799–815. [Google Scholar] [CrossRef]
- Yao, D.M.; Zou, C.; Shu, Y.N.; Liu, S.S. WRKY transcription factors in nicotiana tabacum modulate plant immunity against whitefly via interacting with MAPK cascade pathways. Insects 2020, 12, 16. [Google Scholar] [CrossRef]
- Ohta, M.; Matsui, K.; Hiratsu, K.; Shinshi, H.; Ohme-Takagi, M. Repression domains of class II ERF transcriptional repressors share an essential motif for active repression. Plant Cell 2001, 13, 1959–1968. [Google Scholar] [CrossRef]
- Lorenzo, O.; Piqueras, R.; Sanchez-Serrano, J.J.; Solano, R. ETHYLENE RESPONSE FACTOR1 integrates signals from ethylene and jasmonate pathways in plant defense. Plant Cell 2003, 15, 165–178. [Google Scholar] [CrossRef]
- Lu, J.; Ju, H.P.; Zhou, G.X.; Zhu, C.S.; Erb, M.; Wang, X.P.; Wang, P.; Lou, Y.G. An EAR-motif-containing ERF transcription factor affects herbivore-induced signaling, defense and resistance in rice. Plant J. 2011, 68, 583–596. [Google Scholar] [CrossRef]
- Son, G.H.; Wan, J.R.; Kim, H.J.; Nguyen, X.C.; Chung, W.S.; Hong, J.C.; Stacey, G. Ethylene-responsive element-binding factor 5, ERF5, is involved in chitin-induced innate immunity response. Mol. Plant Microbe Interact. 2012, 25, 48–60. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Reads | Raw Base (G) 1 | Clean Base (G) 2 | Error (%) 3 | Q20 (%) 4 | Q30 (%) 5 | GC Content (%) |
|---|---|---|---|---|---|---|---|---|
| NI-1 | 22825325 | 22389877 | 6.85 | 6.72 | 0.03 | 97.32 | 92.53 | 41.69 |
| NI-2 | 26091282 | 24468330 | 7.83 | 7.34 | 0.03 | 97.89 | 93.69 | 43.02 |
| NI-3 | 28556897 | 26626176 | 8.57 | 7.99 | 0.03 | 97.89 | 93.76 | 43.18 |
| I-12 h-1 | 27446453 | 25817532 | 8.23 | 7.75 | 0.03 | 97.97 | 93.88 | 43.32 |
| I-12 h-2 | 26080109 | 24327843 | 7.82 | 7.3 | 0.03 | 97.95 | 93.89 | 43.31 |
| I-12 h-3 | 26357914 | 24481241 | 7.91 | 7.34 | 0.03 | 97.98 | 93.98 | 43.36 |
| I-24 h-1 | 26781167 | 24971508 | 8.03 | 7.49 | 0.03 | 97.86 | 93.69 | 42.38 |
| I-24 h-2 | 23722409 | 23321513 | 7.12 | 7 | 0.03 | 97.23 | 92.28 | 41.91 |
| I-24 h-3 | 24041972 | 22384567 | 7.21 | 6.72 | 0.03 | 97.91 | 93.79 | 41.82 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Ye, Z.-X.; Wang, Y.-Z.; Wang, X.-J.; Chen, J.-P.; Huang, H.-J. Transcriptomic Analysis of Tobacco Plants in Response to Whitefly Infection. Genes 2023, 14, 1640. https://doi.org/10.3390/genes14081640
Wang X, Ye Z-X, Wang Y-Z, Wang X-J, Chen J-P, Huang H-J. Transcriptomic Analysis of Tobacco Plants in Response to Whitefly Infection. Genes. 2023; 14(8):1640. https://doi.org/10.3390/genes14081640
Chicago/Turabian StyleWang, Xin, Zhuang-Xin Ye, Yi-Zhe Wang, Xiao-Jing Wang, Jian-Ping Chen, and Hai-Jian Huang. 2023. "Transcriptomic Analysis of Tobacco Plants in Response to Whitefly Infection" Genes 14, no. 8: 1640. https://doi.org/10.3390/genes14081640
APA StyleWang, X., Ye, Z.-X., Wang, Y.-Z., Wang, X.-J., Chen, J.-P., & Huang, H.-J. (2023). Transcriptomic Analysis of Tobacco Plants in Response to Whitefly Infection. Genes, 14(8), 1640. https://doi.org/10.3390/genes14081640