
Novel α-1,3-Glucosyltransferase Variants and Their Broad Clinical Polycystic Liver Disease Spectrum

 , , and
, , and
Abstract
1. Introduction

{kind=link}
{kind=link}
{kind=link}
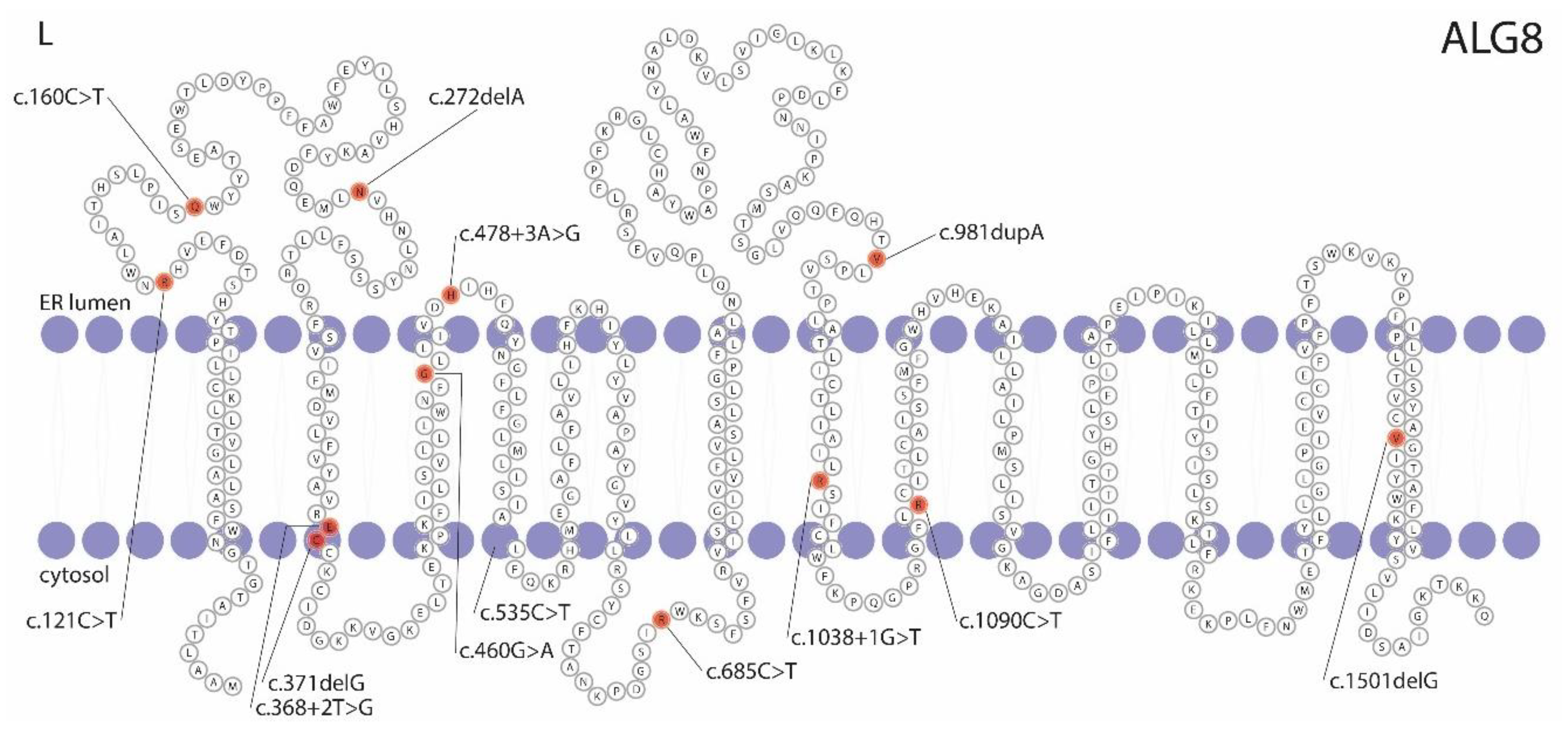{kind=link}
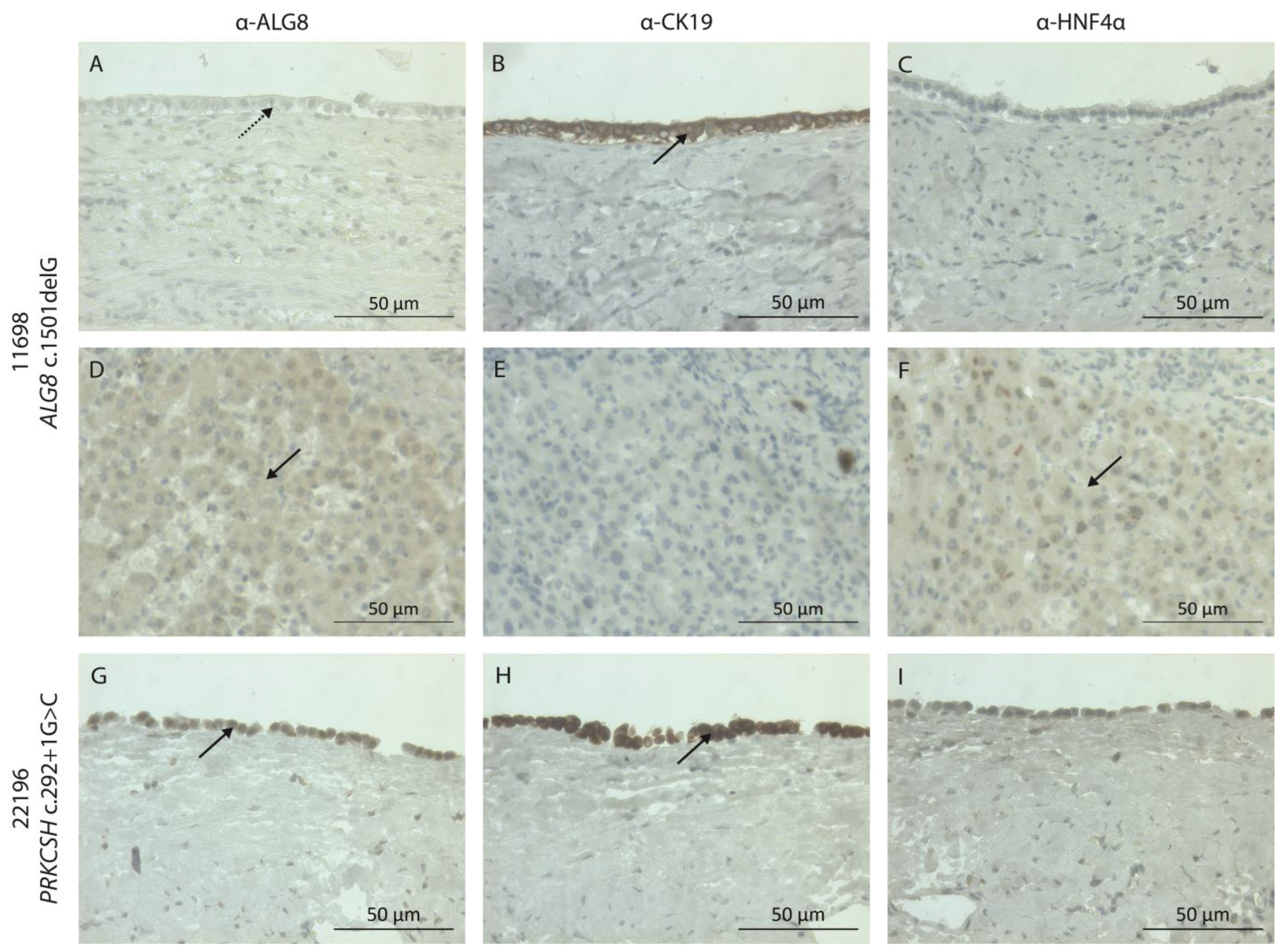{kind=link}
| Gene | Protein | Associated Disease | ||
|---|---|---|---|---|
| Abbreviation | Full Name (NCBI ID) | Abbreviation | Full Name (UniProt ID) | |
| ALG5 | ALG5 dolichyl-phosphate β-glucosyltransferase (29880) | ALG5 | Dolichyl-phosphate β-glucosyltransferase (Q9Y673) | ADPKD [27] |
| ALG8 | ALG8 α-1,3-glucosyltransferase (79053) | ALG8 | Probable dolichyl pyrophosphate Glc1Man9GlcNAc2 α-1,3-glucosyltransferase (Q9BVK2) | ADPLD [36,38] |
| ALG9 | ALG9 α-1,2-mannosyltransferase (79796) | ALG9 | α-1,2-mannosyltransferase (Q9H6U8) | ADPKD and ADPLD [41,42,43] |
| DNAJB11 | DnaJ heat shock protein family (Hsp40) member B11 (51726) | DJB11 or ERJ3 | DnaJ homolog subfamily B member 11 (Q9UBS4) | ADPKD [44,45,46] |
| GANAB | Glucosidase II α subunit (23193) | GANAB or G2AN | Neutral α-glucosidase AB (Q14697) | ADPKD and ADPLD [36,47,48,49,50,51] |
| IFT140 | Intraflagellar transport 140 (9742) | IF140 | Intraflagellar transport protein 140 homolog (Q96RY7) | ADPKD [28] |
| LRP5 | LDL receptor-related protein 5 (4041) | LRP5 | Low-density lipoprotein receptor-related protein 5 (O75197) | ADPKD and ADPLD [52,53] |
| PKD1 | Polycystin 1, transient receptor potential channel interacting (5310) | PC1 or PKD1 | Polycystin-1 (P98161) | ADPKD [2,3,15] |
| PKD2 | Polycystin 2, transient receptor potential cation channel (5311) | PC2 or PKD2 | Polycystin-2 (Q13563) | ADPKD [2,3,15] |
| PKHD1 | PKHD1 ciliary IPT domain containing fibrocystin/ polyductin (5314) | FC or PKHD1 | Fibrocystin (P08F94) | ADPLD [36,54] |
| PRKCSH | Protein kinase C substrate 80K-H (5589) | GLU2B | Glucosidase 2 subunit β (P14314) | ADPLD [2,3,15] |
| SEC61A1 | SEC61 translocon subunit α 1 (29927) | S61A1 | Protein transport protein Sec61 subunit α isoform 1 (P61619) | ADPKD and ADPLD [29] |
| SEC61B | SEC61 translocon subunit β (10952) | SEC61β or SC61B | Protein transport protein Sec61 subunit β (P60468) | ADPLD [36] |
| SEC63 | SEC63 homolog, protein translocation regulator (11231) | SEC63 | Translocation protein SEC63 homolog (Q9UGP8) | ADPLD [2,3,15,51] |
2. Material and Methods
2.1. Cohort Selection
2.2. Genetic Screening
2.3. 3D Modeling
2.4. Immunohistochemistry Staining
3. Results
3.1. ALG8 Variants in the ADPLD Cohort
3.2. The Hepatic Phenotype of ALG8 Patients
3.2.1. Family 1
3.2.2. Family 2
3.2.3. Singletons
3.3. Extrahepatic Manifestations in ALG8 Patients
3.4. Pathogenicity Prediction of ALG8 Variants
3.5. Somatic ALG8 Loss of Heterozygosity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Aerts, R.M.M.; van de Laarschot, L.F.M.; Banales, J.M.; Drenth, J.P.H. Clinical management of polycystic liver disease. J. Hepatol. 2018, 68, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Masyuk, T.V.; Masyuk, A.I.; LaRusso, N.F. Polycystic Liver Disease: Advances in Understanding and Treatment. Annu. Rev. Pathol. 2022, 17, 251–269. [Google Scholar] [CrossRef] [PubMed]
- Olaizola, P.; Rodrigues, P.M.; Caballero-Camino, F.J.; Izquierdo-Sanchez, L.; Aspichueta, P.; Bujanda, L.; Larusso, N.F.; Drenth, J.P.H.; Perugorria, M.J.; Banales, J.M. Genetics, pathobiology and therapeutic opportunities of polycystic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 585–604. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Hu, F.; Ge, X.; Lei, J.; Yu, S.; Wang, T.; Zhou, Q.; Mei, C.; Shi, Y. Structure of the human PKD1-PKD2 complex. Science 2018, 361, eaat9819. [Google Scholar] [CrossRef]
- Brill, A.L.; Ehrlich, B.E. Polycystin 2: A calcium channel, channel partner, and regulator of calcium homeostasis in ADPKD. Cell. Signal. 2020, 66, 109490. [Google Scholar] [CrossRef]
- Padhy, B.; Xie, J.; Wang, R.; Lin, F.; Huang, C.-L. Channel Function of Polycystin-2 in the Endoplasmic Reticulum Protects against Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2022, 33, 1501–1516. [Google Scholar] [CrossRef]
- Hardy, E.; Tsiokas, L. Polycystins as components of large multiprotein complexes of polycystin interactors. Cell. Signal. 2020, 72, 109640. [Google Scholar] [CrossRef]
- Masyuk, T.; Masyuk, A.; LaRusso, N. Polycystic Liver Diseases: Genetics, Mechanisms and Therapies. In The Liver; Wiley-Blackwell: Hoboken, NJ, USA, 2020; pp. 408–421. [Google Scholar]
- Norcia, L.F.; Watanabe, E.M.; Filho, P.T.H.; Hasimoto, C.N.; Pelafsky, L.; de Oliveira, W.K.; Sassaki, L.Y. Polycystic Liver Disease: Pathophysiology, Diagnosis and Treatment. Hepat. Med. 2022, 14, 135–161. [Google Scholar] [CrossRef]
- Yu, Z.; Shen, X.; Hu, C.; Zeng, J.; Wang, A.; Chen, J. Molecular Mechanisms of Isolated Polycystic Liver Diseases. Front. Genet. 2022, 13, 846877. [Google Scholar] [CrossRef]
- Reiterová, J.; Tesař, V. Autosomal Dominant Polycystic Kidney Disease: From Pathophysiology of Cystogenesis to Advances in the Treatment. Int. J. Mol. Sci. 2022, 23, 3317. [Google Scholar] [CrossRef]
- Janssen, M.J.; Waanders, E.; Te Morsche, R.H.; Xing, R.; Dijkman, H.B.; Woudenberg, J.; Drenth, J.P. Secondary, somatic mutations might promote cyst formation in patients with autosomal dominant polycystic liver disease. Gastroenterology 2011, 141, 2056–2063. [Google Scholar] [CrossRef] [PubMed]
- Wills, E.S.; Cnossen, W.R.; Veltman, J.A.; Woestenenk, R.; Steehouwer, M.; Salomon, J.; Te Morsche, R.H.; Huch, M.; Hehir-Kwa, J.Y.; Banning, M.J.; et al. Chromosomal abnormalities in hepatic cysts point to novel polycystic liver disease genes. Eur. J. Hum. Genet. 2016, 24, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Perugorria, M.J.; Banales, J.M. Genetics: Novel causative genes for polycystic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 391–392. [Google Scholar] [CrossRef] [PubMed]
- Boerrigter, M.M.; Bongers, E.; Lugtenberg, D.; Nevens, F.; Drenth, J.P.H. Polycystic liver disease genes: Practical considerations for genetic testing. Eur. J. Med. Genet. 2021, 64, 104160. [Google Scholar] [CrossRef] [PubMed]
- Chandok, N. Polycystic liver disease: A clinical review. Ann. Hepatol. 2012, 11, 819–826. [Google Scholar] [CrossRef]
- Larusso, N.F.; Masyuk, T.V.; Hogan, M.C. Polycystic Liver Disease: The Benefits of Targeting cAMP. Clin. Gastroenterol. Hepatol. 2016, 14, 1031–1034. [Google Scholar] [CrossRef]
- Aapkes, S.E.; Bernts, L.H.P.; Barten, T.R.M.; van den Berg, M.; Gansevoort, R.T.; Drenth, J.P.H. Estrogens in polycystic liver disease: A target for future therapies? Liver Int. 2021, 41, 2009–2019. [Google Scholar] [CrossRef]
- Perugorria, M.J.; Labiano, I.; Esparza-Baquer, A.; Marzioni, M.; Marin, J.J.G.; Bujanda, L.; Banales, J.M. Bile Acids in Polycystic Liver Diseases: Triggers of Disease Progression and Potential Solution for Treatment. Dig. Dis. 2017, 35, 275–281. [Google Scholar] [CrossRef]
- Urribarri, A.D.; Munoz-Garrido, P.; Perugorria, M.J.; Erice, O.; Merino-Azpitarte, M.; Arbelaiz, A.; Lozano, E.; Hijona, E.; Jiménez-Agüero, R.; Fernandez-Barrena, M.G.; et al. Inhibition of metalloprotease hyperactivity in cystic cholangiocytes halts the development of polycystic liver diseases. Gut 2014, 63, 1658–1667. [Google Scholar] [CrossRef]
- Zheng, Q.; Reid, G.; Eccles, M.R.; Stayner, C. Non-coding RNAs as potential biomarkers and therapeutic targets in polycystic kidney disease. Front. Physiol. 2022, 13, 1006427. [Google Scholar] [CrossRef]
- Lee-Law, P.Y.; Olaizola, P.; Caballero-Camino, F.J.; Izquierdo-Sanchez, L.; Rodrigues, P.M.; Perugorria, M.J.; Azkargorta, M.; Elortza, F.; Martinez-Chantar, M.L.; Aspichueta, P.; et al. Inhibition of NAE-dependent protein hyper-NEDDylation in cystic cholangiocytes halts cystogenesis in experimental models of polycystic liver disease. United Eur. Gastroenterol. J. 2021, 9, 848–859. [Google Scholar] [CrossRef]
- Lee-Law, P.Y.; Olaizola, P.; Caballero-Camino, F.J.; Izquierdo-Sanchez, L.; Rodrigues, P.M.; Santos-Laso, A.; Azkargorta, M.; Elortza, F.; Martinez-Chantar, M.L.; Perugorria, M.J.; et al. Targeting UBC9-mediated protein hyper-SUMOylation in cystic cholangiocytes halts polycystic liver disease in experimental models. J. Hepatol. 2021, 74, 394–406. [Google Scholar] [CrossRef] [PubMed]
- Masyuk, A.I.; Masyuk, T.V.; Pisarello, M.J.L.; Ding, J.; Loarca, L.; Huang, B.Q.; LaRusso, N.F. Cholangiocyte autophagy contributes to hepatic cystogenesis in polycystic liver disease and represents a potential therapeutic target. Hepatology 2018, 67, 1088–1108. [Google Scholar] [CrossRef]
- Santos-Laso, A.; Izquierdo-Sanchez, L.; Rodrigues, P.M.; Huang, B.Q.; Azkargorta, M.; Lapitz, A.; Munoz-Garrido, P.; Arbelaiz, A.; Caballero-Camino, F.J.; Fernández-Barrena, M.G.; et al. Proteostasis disturbances and endoplasmic reticulum stress contribute to polycystic liver disease: New therapeutic targets. Liver Int. 2020, 40, 1670–1685. [Google Scholar] [CrossRef]
- Li, X. Epigenetics and cell cycle regulation in cystogenesis. Cell. Signal. 2020, 68, 109509. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, H.; Raud, L.; Foulquier, F.; Sayer, J.A.; Lambert, B.; Olinger, E.; Lefèvre, S.; Knebelmann, B.; Harris, P.C.; Trouvé, P.; et al. Monoallelic pathogenic ALG5 variants cause atypical polycystic kidney disease and interstitial fibrosis. Am. J. Hum. Genet. 2022, 109, 1484–1499. [Google Scholar] [CrossRef] [PubMed]
- Senum, S.R.; Li, Y.S.M.; Benson, K.A.; Joli, G.; Olinger, E.; Lavu, S.; Madsen, C.D.; Gregory, A.V.; Neatu, R.; Kline, T.L.; et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am. J. Hum. Genet. 2022, 109, 136–156. [Google Scholar] [CrossRef] [PubMed]
- Schlevogt, B.; Schlieper, V.; Krader, J.; Schröter, R.; Wagner, T.; Weiand, M.; Zibert, A.; Schmidt, H.H.; Bergmann, C.; Nedvetsky, P.I.; et al. A SEC61A1 variant is associated with autosomal dominant polycystic liver disease. Liver Int. 2023, 43, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Lanktree, M.B.; Iliuta, I.-A.; Haghighi, A.; Song, X.; Pei, Y. Evolving role of genetic testing for the clinical management of autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 2018, 34, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, T.; Teraoka, A.; Akagawa, H.; Makabe, S.; Akihisa, T.; Sato, M.; Kataoka, H.; Mitobe, M.; Furukawa, T.; Tsuchiya, K.; et al. Mutation analyses by next-generation sequencing and multiplex ligation-dependent probe amplification in Japanese autosomal dominant polycystic kidney disease patients. Clin. Exp. Nephrol. 2019, 23, 1022–1030. [Google Scholar] [CrossRef]
- Mallawaarachchi, A.C.; Lundie, B.; Hort, Y.; Schonrock, N.; Senum, S.R.; Gayevskiy, V.; Minoche, A.E.; Hollway, G.; Ohnesorg, T.; Hinchcliffe, M.; et al. Genomic diagnostics in polycystic kidney disease: An assessment of real-world use of whole-genome sequencing. Eur. J. Hum. Genet. 2021, 29, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Aebi, M. N-linked protein glycosylation in the ER. Biochim. Biophys. Acta 2013, 1833, 2430–2437. [Google Scholar] [CrossRef]
- Höck, M.; Wegleiter, K.; Ralser, E.; Kiechl-Kohlendorfer, U.; Scholl-Bürgi, S.; Fauth, C.; Steichen, E.; Pichler, K.; Lefeber, D.J.; Matthjis, G.; et al. ALG8-CDG: Novel patients and review of the literature. Orphanet J. Rare Dis. 2015, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Albokhari, D.; Ng, B.G.; Guberinic, A.; Daniel, E.J.P.; Engelhardt, N.M.; Barone, R.; Fiumara, A.; Garavelli, L.; Trimarchi, G.; Wolfe, L.; et al. ALG8-CDG: Molecular and phenotypic expansion suggests clinical management guidelines. J. Inherit. Metab. Dis. 2022, 45, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Besse, W.; Dong, K.; Choi, J.; Punia, S.; Fedeles, S.V.; Choi, M.; Gallagher, A.R.; Huang, E.B.; Gulati, A.; Knight, J.; et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J. Clin. Investig. 2017, 127, 1772–1785. [Google Scholar] [CrossRef]
- Lanktree, M.B.; Haghighi, A.; Guiard, E.; Iliuta, I.A.; Song, X.; Harris, P.C.; Paterson, A.D.; Pei, Y. Prevalence Estimates of Polycystic Kidney and Liver Disease by Population Sequencing. J. Am. Soc. Nephrol. 2018, 29, 2593–2600. [Google Scholar] [CrossRef]
- Apple, B.; Sartori, G.; Moore, B.; Chintam, K.; Singh, G.; Anand, P.M.; Strande, N.; Mirshahi, T.; Triffo, W.; Chang, A.R. Individuals heterozygous for ALG8 protein-truncating variants are at increased risk of a mild cystic kidney disease. Kidney Int. 2022, 103, 607–615. [Google Scholar] [CrossRef]
- Suwabe, T.; Shukoor, S.; Chamberlain, A.M.; Killian, J.M.; King, B.F.; Edwards, M.; Senum, S.R.; Madsen, C.D.; Chebib, F.T.; Hogan, M.C.; et al. Epidemiology of Autosomal Dominant Polycystic Kidney Disease in Olmsted County. Clin. J. Am. Soc. Nephrol. 2020, 15, 69–79. [Google Scholar] [CrossRef]
- Suwabe, T.; Chamberlain, A.M.; Killian, J.M.; King, B.F.; Gregory, A.V.; Madsen, C.D.; Wang, X.; Kline, T.L.; Chebib, F.T.; Hogan, M.C.; et al. Epidemiology of autosomal-dominant polycystic liver disease in Olmsted county. JHEP Rep. 2020, 2, 100166. [Google Scholar] [CrossRef]
- Besse, W.; Chang, A.R.; Luo, J.Z.; Triffo, W.J.; Moore, B.S.; Gulati, A.; Hartzel, D.N.; Mane, S.; Center, R.G.; Torres, V.E.; et al. ALG9 Mutation Carriers Develop Kidney and Liver Cysts. J. Am. Soc. Nephrol. 2019, 30, 2091–2102. [Google Scholar] [CrossRef]
- Schönauer, R.; Baatz, S.; Nemitz-Kliemchen, M.; Frank, V.; Petzold, F.; Sewerin, S.; Popp, B.; Münch, J.; Neuber, S.; Bergmann, C.; et al. Matching clinical and genetic diagnoses in autosomal dominant polycystic kidney disease reveals novel phenocopies and potential candidate genes. Genet. Med. 2020, 22, 1374–1383. [Google Scholar] [CrossRef] [PubMed]
- Boerrigter, M.M.; Duijzer, R.; te Morsche, R.H.M.; Drenth, J.P.H. Heterozygosity of ALG9 in association with autosomal dominant polycystic liver disease. Am. J. Hum. Genet. 2023, 102, 832–844. [Google Scholar]
- Cornec-Le Gall, E.; Olson, R.J.; Besse, W.; Heyer, C.M.; Gainullin, V.G.; Smith, J.M.; Audrézet, M.P.; Hopp, K.; Porath, B.; Shi, B.; et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am. J. Hum. Genet. 2018, 102, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Pisani, I.; Allinovi, M.; Palazzo, V.; Zanelli, P.; Gentile, M.; Farina, M.T.; Giuliotti, S.; Cravedi, P.; Delsante, M.; Maggiore, U.; et al. More dissimilarities than affinities between DNAJB11-PKD and ADPKD. Clin. Kidney J. 2022, 15, 1179–1187. [Google Scholar] [CrossRef]
- Huynh, V.T.; Audrézet, M.P.; Sayer, J.A.; Ong, A.C.; Lefevre, S.; Le Brun, V.; Després, A.; Senum, S.R.; Chebib, F.T.; Barroso-Gil, M.; et al. Clinical spectrum, prognosis and estimated prevalence of DNAJB11-kidney disease. Kidney Int. 2020, 98, 476–487. [Google Scholar] [CrossRef]
- Van de Laarschot, L.F.M.; Te Morsche, R.H.M.; Hoischen, A.; Venselaar, H.; Roelofs, H.M.; Cnossen, W.R.; Banales, J.M.; Roepman, R.; Drenth, J.P.H. Novel GANAB variants associated with polycystic liver disease. Orphanet J. Rare Dis. 2020, 15, 302. [Google Scholar] [CrossRef] [PubMed]
- Porath, B.; Gainullin, V.G.; Cornec-Le Gall, E.; Dillinger, E.K.; Heyer, C.M.; Hopp, K.; Edwards, M.E.; Madsen, C.D.; Mauritz, S.R.; Banks, C.J.; et al. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am. J. Hum. Genet. 2016, 98, 1193–1207. [Google Scholar] [CrossRef]
- Delbarba, E.; Econimo, L.; Dordoni, C.; Martin, E.; Mazza, C.; Savoldi, G.; Alberici, F.; Scolari, F.; Izzi, C. Expanding the variability of the ADPKD-GANAB clinical phenotype in a family of Italian ancestry. J. Nephrol. 2022, 35, 645–652. [Google Scholar] [CrossRef]
- Besse, W.; Choi, J.; Ahram, D.; Mane, S.; Sanna-Cherchi, S.; Torres, V.; Somlo, S. A noncoding variant in GANAB explains isolated polycystic liver disease (PCLD) in a large family. Hum. Mutat. 2018, 39, 378–382. [Google Scholar] [CrossRef]
- Wilson, E.M.; Choi, J.; Torres, V.E.; Somlo, S.; Besse, W. Large Deletions in GANAB and SEC63 Explain 2 Cases of Polycystic Kidney and Liver Disease. Kidney Int. Rep. 2020, 5, 727–731. [Google Scholar] [CrossRef]
- Cnossen, W.R.; te Morsche, R.H.M.; Hoischen, A.; Gilissen, C.; Chrispijn, M.; Venselaar, H.; Mehdi, S.; Bergmann, C.; Veltman, J.A.; Drenth, J.P.H. Whole-exome sequencing reveals LRP5 mutations and canonical Wnt signaling associated with hepatic cystogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 5343–5348. [Google Scholar] [CrossRef] [PubMed]
- Cnossen, W.R.; te Morsche, R.H.M.; Hoischen, A.; Gilissen, C.; Venselaar, H.; Mehdi, S.; Bergmann, C.; Losekoot, M.; Breuning, M.H.; Peters, D.J.M.; et al. LRP5 variants may contribute to ADPKD. Eur. J. Hum. Genet. 2016, 24, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, H.; Guo, R.; Sang, X.; Mao, Y. Association of a novel PKHD1 mutation in a family with autosomal dominant polycystic liver disease. Ann. Transl. Med. 2021, 9, 120. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2022, 51, D523–D531. [Google Scholar] [CrossRef]
- D’Agnolo, H.M.; Kievit, W.; Andrade, R.J.; Karlsen, T.H.; Wedemeyer, H.; Drenth, J.P. Creating an effective clinical registry for rare diseases. United Eur. Gastroenterol. J. 2016, 4, 333–338. [Google Scholar] [CrossRef]
- Ravine, D.; Sheffield, L.; Danks, D.; Gibson, R.N.; Walker, R.; Kincaid-Smith, P. Evaluation of ultrasonographic diagnostic criteria for autosomal dominant polycystic kidney disease 1. Lancet 1994, 343, 824–827. [Google Scholar] [CrossRef]
- Pei, Y.; Obaji, J.; Dupuis, A.; Paterson, A.D.; Magistroni, R.; Dicks, E.; Parfrey, P.; Cramer, B.; Coto, E.; Torra, R.; et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J. Am. Soc. Nephrol. 2009, 20, 205–212. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. Available online: http://evs.gs.washington.edu/EVS/) (accessed on 1 June 2022).
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2021, 50, D439–D444. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Albuquerque-Wendt, A.; Hütte, H.J.; Buettner, F.F.R.; Routier, F.H.; Bakker, H. Membrane Topological Model of Glycosyltransferases of the GT-C Superfamily. Int. J. Mol. Sci. 2019, 20, 4842. [Google Scholar] [CrossRef] [PubMed]
- Barten, T.R.M.; Bökkerink, R.M.P.; Venderink, W.; Gevers, T.J.G.; Broek, R.P.G.T.; Drenth, J.P.H. Abdominal wall hernia is a frequent complication of polycystic liver disease and associated with hepatomegaly. Liver Int. 2022, 42, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, V.; Bin, S.; Graziano, C.; Capelli, I.; Minardi, R.; Aiello, V.; Ambrosini, E.; Cristalli, C.P.; Mattiaccio, A.; Pariali, M.; et al. Gene Panel Analysis in a Large Cohort of Patients with Autosomal Dominant Polycystic Kidney Disease Allows the Identification of 80 Potentially Causative Novel Variants and the Characterization of a Complex Genetic Architecture in a Subset of Families. Front. Genet. 2020, 11, 464. [Google Scholar] [CrossRef]
- Watnick, T.J.; Torres, V.E.; Gandolph, M.A.; Qian, F.; Onuchic, L.F.; Klinger, K.W.; Landes, G.; Germino, G.G. Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease. Mol. Cell 1998, 2, 247–251. [Google Scholar] [CrossRef]
- Pei, Y.; Watnick, T.; He, N.; Wang, K.; Liang, Y.; Parfrey, P.; Germino, G.; St George-Hyslop, P. Somatic PKD2 mutations in individual kidney and liver cysts support a “two-hit” model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 1999, 10, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Torres, V.E.; Harris, P.C. Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J. Am. Soc. Nephrol. 2018, 29, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Karhunen, P.J.; Tenhu, M. Adult polycystic liver and kidney diseases are separate entities. Clin. Genet. 1986, 30, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Sherstha, R.; McKinley, C.; Russ, P.; Scherzinger, A.; Bronner, T.; Showalter, R.; Everson, G.T. Postmenopausal estrogen therapy selectively stimulates hepatic enlargement in women with autosomal dominant polycystic kidney disease. Hepatology 1997, 26, 1282–1286. [Google Scholar] [CrossRef]
- Ben Ayed, I.; Ouarda, W.; Frikha, F.; Kammoun, F.; Souissi, A.; Ben Said, M.; Bouzid, A.; Elloumi, I.; Hamdani, T.M.; Gharbi, N.; et al. SRD5A3-CDG: 3D structure modeling, clinical spectrum, and computer-based dysmorphic facial recognition. Am. J. Med. Genet. Part A 2021, 185, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Cherepanova, N.; Shrimal, S.; Gilmore, R. N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 2016, 41, 57–65. [Google Scholar] [CrossRef]
- Starosta, R.T.; Boyer, S.; Tahata, S.; Raymond, K.; Lee, H.E.; Wolfe, L.A.; Lam, C.; Edmondson, A.C.; Schwartz, I.V.D.; Morava, E. Liver manifestations in a cohort of 39 patients with congenital disorders of glycosylation: Pin-pointing the characteristics of liver injury and proposing recommendations for follow-up. Orphanet J. Rare Dis. 2021, 16, 20. [Google Scholar] [CrossRef] [PubMed]
- Vaz-Drago, R.; Custódio, N.; Carmo-Fonseca, M. Deep intronic mutations and human disease. Hum. Genet. 2017, 136, 1093–1111. [Google Scholar] [CrossRef]
- Schollen, E.; Keldermans, L.; Foulquier, F.; Briones, P.; Chabas, A.; Sánchez-Valverde, F.; Adamowicz, M.; Pronicka, E.; Wevers, R.; Matthijs, G. Characterization of two unusual truncating PMM2 mutations in two CDG-Ia patients. Mol. Genet. Metab. 2007, 90, 408–413. [Google Scholar] [CrossRef]
- Vega, A.I.; Pérez-Cerdá, C.; Desviat, L.R.; Matthijs, G.; Ugarte, M.; Pérez, B. Functional analysis of three splicing mutations identified in the PMM2 gene: Toward a new therapy for congenital disorder of glycosylation type Ia. Hum. Mutat. 2009, 30, 795–803. [Google Scholar] [CrossRef]
- Michel-Calemard, L.; Dijoud, F.; Till, M.; Lambert, J.C.; Vercherat, M.; Tardy, V.; Coubes, C.; Morel, Y. Pseudoexon activation in the PKHD1 gene: A French founder intronic mutation IVS46+653A>G causing severe autosomal recessive polycystic kidney disease. Clin. Genet. 2009, 75, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ma, N.; Zhao, X.; Li, W.; Zhang, Q.; Yuan, S.; Tan, Y.-Q.; Lu, G.; Lin, G.; Du, J. A rare deep intronic mutation of PKHD1 gene, c.8798-459 C > A, causes autosomal recessive polycystic kidney disease by pseudoexon activation. J. Hum. Genet. 2019, 64, 207–214. [Google Scholar] [CrossRef] [PubMed]





| Chromosome Position | Nucleotide Change | Amino Acid Change | Variant Type | ACMG/AMP | Patient | Sex | Age | Hepatic Cysts | Imaging | GGT | Renal Cysts | eGFR | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| g.78127372G>A | c.160C>T | p.(Gln54*) | Nonsense | Pathogenic | 9960 | Female | 87 | PLD | US | - | - | - | Family 1 |
| 8826 | Female | 60 | 20+ | CT | 44 | 1 | 69 | ||||||
| 9173 | Female | 44 | 20+ | CT | 15 | 1 | 84 | ||||||
| 9244 | Female | 58 | PLD | US | - | - | - | ||||||
| g.78124118del | c.272delA | p.(Asn91Metfs*5) | Frameshift | VUS | 3642 | Male | 29 | 10+ | CT | 17 | 4 | 89 | |
| 7906 | Female | 50 | PLD | US | - | - | - | ||||||
| g.78121172del | c.371delG | p.(Cys124Serfs*33) | Frameshift | Likely pathogenic | 8515 | Male | 75 | 20+ | CT | 762 a | 1 | >90 | |
| g.78121083C>T | c.460G>A | p.(Gly154Arg) | Missense | VUS | 11549 | Female | 73 | PLD | US | - | - | - | |
| g.78121062T>C | c.478+3A>G | p.? | Splice-site | VUS | 10027 | Female | 56 | 10+ | CT | 29 | 1 | 77 | |
| g.78113978G>A | c.685C>T | p.(Arg229*) | Nonsense | Pathogenic | 6935 | Female | 48 | 20+ | CT | 16 | 0 | 84 | Family 2 |
| 24392 | Male | 68 | PLD | US | - | 0 | 86 | ||||||
| g.78109499dup | c.981dupA | p.(Val328Serfs*28) | Frameshift | Pathogenic | 762 | Male | 66 | 5 | CT | 25 | 3 | 79 | |
| g.78106895G>A | c.1090C>T [36,38] | p.(Arg364*) | Nonsense | Pathogenic | 23932 | Female | 59 | 20+ | CT | 410 a | 0 | >90 | |
| 11409 | Female | 86 | PLD | US | - | - | - | ||||||
| g.78101044del | c.1501delG | p.(Val501*) | Nonsense | Likely pathogenic | 11698 | Male | 57 | 20+ | CT | - | 5 | - | |
| 8094 | Female | 48 | PLD | US | - | 7 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boerrigter, M.M.; te Morsche, R.H.M.; Venselaar, H.; Pastoors, N.; Geerts, A.M.; Hoorens, A.; Drenth, J.P.H. Novel α-1,3-Glucosyltransferase Variants and Their Broad Clinical Polycystic Liver Disease Spectrum. Genes 2023, 14, 1652. https://doi.org/10.3390/genes14081652
Boerrigter MM, te Morsche RHM, Venselaar H, Pastoors N, Geerts AM, Hoorens A, Drenth JPH. Novel α-1,3-Glucosyltransferase Variants and Their Broad Clinical Polycystic Liver Disease Spectrum. Genes. 2023; 14(8):1652. https://doi.org/10.3390/genes14081652
Chicago/Turabian StyleBoerrigter, Melissa M., René H. M. te Morsche, Hanka Venselaar, Nikki Pastoors, Anja M. Geerts, Anne Hoorens, and Joost P. H. Drenth. 2023. "Novel α-1,3-Glucosyltransferase Variants and Their Broad Clinical Polycystic Liver Disease Spectrum" Genes 14, no. 8: 1652. https://doi.org/10.3390/genes14081652
APA StyleBoerrigter, M. M., te Morsche, R. H. M., Venselaar, H., Pastoors, N., Geerts, A. M., Hoorens, A., & Drenth, J. P. H. (2023). Novel α-1,3-Glucosyltransferase Variants and Their Broad Clinical Polycystic Liver Disease Spectrum. Genes, 14(8), 1652. https://doi.org/10.3390/genes14081652