Abstract
CASK-related disorders are a form of rare X-linked neurological diseases and most of the patients are females. They are characterized by several symptoms, including microcephaly with pontine and cerebellar hypoplasia (MICPCH), epilepsy, congenital nystagmus, and neurodevelopmental disorders. Whole-genome sequencing has identified various mutations, including nonsense and missense mutations, from patients with CASK-related disorders, revealing correlations between specific mutations and clinical phenotypes. Notably, missense mutations associated with epilepsy and intellectual disability were found throughout the whole region of the CASK protein, while missense mutations related to microcephaly and MICPCH were restricted in certain domains. To investigate the pathophysiology of CASK-related disorders, research groups have employed diverse methods, including the generation of CASK knockout mice and the supplementation of CASK to rescue the phenotypes. These approaches have yielded valuable insights into the identification of functional domains of the CASK protein associated with a specific phenotype. Additionally, recent advancements in the AI-based prediction of protein structure, such as AlphaFold2, and the application of genome-editing techniques to generate CASK mutant mice carrying missense mutations from patients with CASK-related disorders, allow us to understand the pathophysiology of CASK-related disorders in more depth and to develop novel therapeutic methods for the fundamental treatment of CASK-related disorders.
Keywords:
microcephaly with pontine and cerebellar hypoplasia (MICPCH); calcium/calmodulin-dependent serine protein kinase (CASK); CASK-related disorders; developmental epileptic encephalopathy; X chromosome inactivation; X-linked intellectual developmental disorders with nystagmus; neurocircuit interference 1. CASK-Related Disorders, X-Linked Neurological Disorders
Calcium/calmodulin-dependent serine protein kinase (CASK)-related disorders are a syndrome of multiple pathologies that occur mostly in the nervous system. The CASK gene is located on the X chromosome at Xp11.4 [1,2] and several clinical phenotypes of CASK-related disorders have been identified. Historically, Dimitratos et al. first reported the association between the CASK gene and the clinical phenotype in 1998 [2]. They reported that a deletion of Xp11.4, which contains the CASK gene, was found in patients with X-linked optic atrophy. In 2007, a case of a female patient with a relatively large genomic deletion containing the CASK gene was reported by Froyen et al. [3]. This patient exhibited microcephaly as well as intellectual disability, suggesting that the CASK gene may be involved in X-linked intellectual disability (XLID). The genomic deletions described in this report include genes other than the CASK gene, and it was not concluded that the CASK gene was the cause of microcephaly and XLID. In 2008, Najm et al. further confirmed the association between CASK gene mutations and microcephaly with cerebellar and pontine hypoplasia (MICPCH) and XLID in five patients [4]. Subsequently, epilepsy syndromes occurring in neonates and infants were also reported by several research groups [5,6,7,8,9,10]. To date, CASK-related disorders comprise various developmental disorders such as MICPCH [4,5], X-linked intellectual disability [7,11], FG syndrome [12,13], pediatric epilepsy syndrome [5,6,7,8,9,10], ophthalmologic phenotypes [13,14] (such as nystagmus), hearing impairment [5,7,11], and autism spectrum disorders [15]. Although CASK is known to be involved in other biological aspects such as cardiogenesis [16] and tumorigenesis [17,18,19,20], this review will focus on the biological role of CASK in the nervous system and the pathophysiology of CASK-related disorders.
2. Protein Structure and Protein Interaction of CASK
CASK was originally identified as an intracellular binding partner of neurexin, a synaptic adhesion molecule [21]. The CASK gene encodes a multiple-domain scaffolding protein, which consists of a catalytic Serine/Threonine Kinase (aka calmodulin kinase (CaMK)) domain, two Lin2/Lin7 (L27.1 and L27.2) domains, a PSD-95/discs large/ZO-1 (PDZ) domain, an Src Homology 3 (SH3) domain, and a Guanylate Kinase (GuK) domain, and each domain plays different roles through their interactions with other proteins [21,22,23].
After neurexin was identified as a binding partner of CASK, more than 50 proteins were demonstrated to bind to CASK (Figure 1A). Biochemical studies have determined the functional domains of CASK recruited in protein–protein interactions (Figure 1B). The CaMK domain at the N-terminal of CASK functions as a magnesium-dependent atypical kinase, which could phosphorylate the C-terminal of neurexins [24]. The CaMK domain has also been demonstrated to interact with other molecules such as liprin-α and Mint1 [25,26,27,28,29]. The L27 domain interacts with the Lin-7 (Veli) family [22,23] and SAP-97 (DLG1) [27,30,31], a member of PSD-95-like membrane-associated guanylate kinases. Both lin-7 and SAP-97 bind to the L27 domains of CASK through its L27 domain on the N-terminal and C-terminal, respectively. SAP97 and Lin-7 are also scaffolding proteins and assemble synaptic proteins. CASK and SAP-97 have been shown to mediate the sorting of the N-methyl-D-aspartate (NMDA) receptor and CASK and Lin-7 regulate the localization of the inward rectifier potassium channel Kir2.3 on the basolateral membrane [32,33]. The PDZ domain of CASK has been shown to bind to trans-membrane proteins including neurexins and syndecans [1,34,35]. Neurexins are presynaptic adhesion proteins that bind to postsynaptic adhesion molecules such as neuroligins and the leucine-rich repeat trans-membrane neuronal proteins (LRRTMs) to construct and maintain the synaptic structure and function [36]. The SH3 domain of CASK binds to the proline-rich region at the carboxyl terminal of the N-type calcium channel and may regulate synaptic vesicle exocytosis at presynaptic axon terminals [37,38]. The GuK domain located at the C-terminal of CASK has been shown to bind to transcription factors, TBR-1, and CASK-interacting Nucleosome Assembly Protein (CINAP) [39,40,41]. The complex of CASK and TBR1 is trans-located in the nuclei of neurons and regulates the expression of genes such as grin2b and reln, which control neuronal migration and maturation [40,42,43].
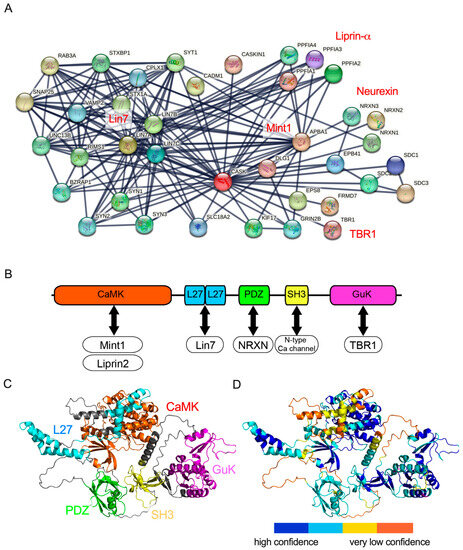
Figure 1.
Protein–protein interaction network and predicted protein structure of CASK. (A) CASK-protein interaction network produced by String database [44]. Protein families that have been intensively investigated are indicated in red. (B) Functional domains of CASK and interactive partner molecules in (A). Five functional domains of CASK, CaMK, L27, PDZ, SH3, and GuK are colored orange, cyan, green, yellow, and magenta, respectively. (C) A three-dimensional structure of CASK is predicted by ColabFold, an AlphaFold2 on Google Colab [45]. (D) The confidence of the prediction is colored based on the score of pLDDT. High-confidence (pLDDT > 90) is drawn in blue and very low confidence is shown in yellow. The angle of the 3D structure of CASK is identical between C and D.
These functional domains of CASK cooperatively bind to partner proteins, enabling CASK to carry out its biological functions. For instance, the PDZ domain of the CASK protein has been recognized as the primary binding domain for neurexin [1,34,35]. However, a recent report of two CASK missense mutations from patients with MICPCH suggested that not only the PDZ domain but also the SH3 and GuK domains of CASK may contribute to the binding between neurexin and CASK [46]. This indicates that the protein interactions between CASK and its partners could be established through multiple domains of the CASK protein.
To understand the macro-view of the protein–protein interaction, it is essential to gain information on the three-dimensional (3D) structures of CASK proteins. Until a few years ago, the 3D structures of each functional domain were demonstrated using X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy, and these data are widely available from the RCSB Protein Data Bank. However, the complete 3D structure of the entire CASK protein remained elusive. Recently, more sophisticated predictions of protein structures were made available using artificial intelligence-based programs, such as AlphaFold [47]. In our investigation, we used ColabFold [45], which is a web-based implementation of AlphaFold2 on Google Collaboratory, to predict the protein structure of CASK (Figure 1C). The prediction achieved high quality in the scale of each functional domain of the CASK protein. The prediction of the entire structure of the CASK protein, however, may not be accurate enough because the model’s confidence is not sufficiently high, especially in the linker regions bridging functional domains of the CASK protein (Figure 1D). Thus, the relative topology of functional domains of the CASK protein may not be precisely predicted by AlfphaFold2. Future improvements in the AI-based prediction will be required to advance our understanding of the biophysical mechanisms of the CASK–protein interactions and to explore potential novel partner proteins binding to CASK.
3. Genetics of CASK-Related Disorders
CASK-related disorders are caused by pathogenic variants in the CASK gene and include a spectrum of clinical phenotypes. CASK-related disorders are classified into two primary phenotypes, MICPCH, and XLID with or without nystagmus. Typically, MICPCH is associated with loss-of-function (i.e., nonsense or frameshift) mutations of the CASK gene, while XLID with or without nystagmus appears to be associated with hypomorphic (i.e., missense) mutations of the CASK gene. We first used the ClinVar database on NCBI [48] and found a total of 306 variants, consisting of 37 frameshift variants (two cases with substitution with a stop codon), 227 missense variants, and 43 nonsense variants (Table 1). Almost all the frameshift and nonsense variants are classified as pathogenic or likely pathogenic, whereas some of the missense variants are classified as benign, indicating that frameshift and nonsense mutations may cause a severer phenotype than missense mutations (p < 0.0001, Chi-square test). However, some variants are not associated with clinical phenotypes on the database (12/37 in frameshift; 58/227 in missense; and 15/44 in nonsense). To evaluate the association between the mutations of the CASK gene and clinical phenotypes, we investigated 49 reports describing 197 patients with CASK-related disorders [4,5,6,7,8,9,10,11,12,13,14,46,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85] and classified the cases based on the genders and phenotypes (Table 2).

Table 1.
Genetic variants in the CASK protein on ClinVar database.
Table 2.
Gender and phenotypes in CASK-related disorders.
Intellectual disability (ID) is been commonly observed both in males (96.1%) and females (93.5%; p = 0.4315, males vs. females; statistically not significant between gender, Chi-square test) affected by CASK-related disorders. MICPCH is relatively common in patients with CASK-related disorders at a lower frequency, both in males (76.0%) and females (87.7%; p = 0.0634, males vs. females; statistically not significant between gender, Chi-square test). Another prominent phenotype associated with CASK-related disorders is epileptic syndromes, encompassing various types of epileptic seizures, including Ohtahara syndrome (aka epileptic encephalopathy with suppression burst of spikes), West syndrome (aka infantile spasms), absence epilepsy, myoclonic seizures, and focal seizures. Epileptic phenotypes were reported in males (54.1%) more frequently than in females (36.1%, p = 0.0198, males vs. females; statistically significant p < 0.05, Chi-square test). Giacomini et al. investigated epileptic phenotypes of 34 Italian patients with CASK-related disorders and reported that half of the patients (50%, 17/34) showed epileptic EEG patterns. Considering these findings together, it is evident that approximately half of the patients suffered from epileptic seizures.
The relationship between clinical phenotypes and types of mutations in the CASK gene shows a sexual difference (Table 3 and Figure 2). Specifically, nonsense mutations, including frameshift mutations, are more prevalent in females than in males. In females, more than 85% of the cases are due to nonsense mutations of the CASK gene, whereas 38~63% of the cases are caused by nonsense mutations in males (p < 0.0001, intellectual disability; p < 0.0001, MICPCH; p < 0.0001, epilepsy; p = 0.0169, opthalmological anomalies; statistically significant in all cases, see Figure 2).
Table 3.
Clinical phenotypes and mutations in CASK-related disorders.
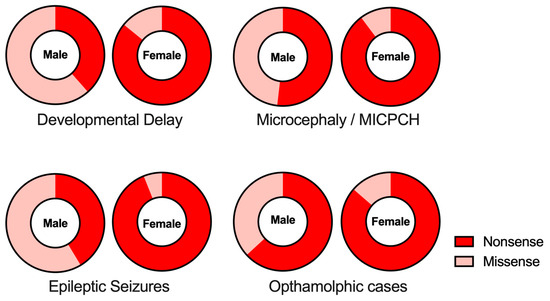
Figure 2.
Gender-dependency of CASK mutation types in clinical phenotypes. The proportions of nonsense (red) and missense (pink) mutations found in clinical phenotypes are shown in each gender.
In CASK-related disorders, nonsense and frameshift mutations are speculated to lead to loss-of-function variants, while missense mutations to hypomorphic variants. Although there are reports that missense or nonsense mutations could become gain-of-function in other disorders (e.g., NLGN3R451C missense mutation [86,87], and SIK1Q633X nonsense mutation [88,89]), to our knowledge, none of the nonsense and missense variants of the CASK protein were demonstrated to be gain-of-function or dominant-negative mutations according to biochemical analysis. The difference in effects between genders may be due to the regulation of the CASK gene by X chromosome inactivation (XCI) [90,91,92]. In males, a mutation in the X chromosome affects all somatic cells. In females, XCI silences one X chromosome, resulting in two cell types: those with CASK variants and those with normal CASK protein (Figure 3A). This could explain how normal cells in females mitigate the impact of loss-of-function CASK variants and how male patients with loss-of-function mutations exhibit severer symptoms, potentially leading to early lethality.
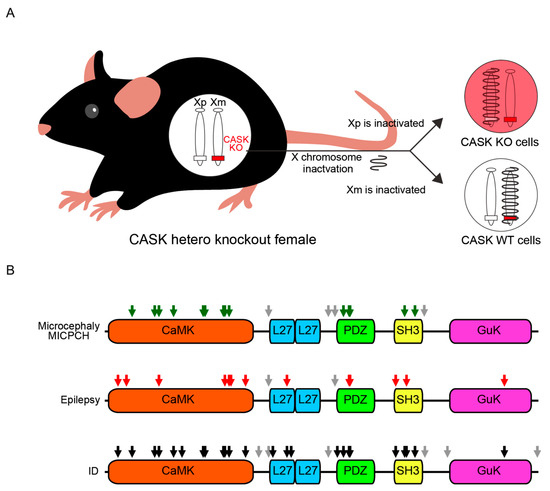
Figure 3.
X chromosome inactivation of the CASK gene and the genomic locus of the missense mutations associated with specific clinical phenotypes. (A) A schematic drawing of X chromosome inactivation (XCI) of the CASK gene in CASK-related disorders. Female mice, as well as human patients, have two X chromosomes, and one of them carries a non-functional/hypomorphic cask gene. One of the two X chromosomes is inactivated and the cells with non-functional (red) and functional (white) cask are generated. (B) A schematic drawing of functional domains of CASK protein and the locations of missense mutations. Grey arrows represent missense mutations outside the five functional domains. Green arrows (top) are the missense mutation identified from patients with MICPCH, red arrows are those with epilepsy, and black arrows are those with ID.
4. Phenotypes and Functional Domains of CASK
Most nonsense and frameshift mutations are considered to result in the degeneration of the protein, mainly by nonsense-mediated mRNA decay (NMD) [93]. NMD is one of the mRNA quality-control mechanisms of eukaryotes. When an immature stop codon is created in the translation region by the nonsense or frameshift mutations, the abnormal mRNA is degraded and almost no protein is translated. On the other hand, it is known that missense mutations can produce the full length of the protein with hypomorphic functions. This idea seems to be supported by the data of ClinVar, showing that more variants classified as the clinical significance of benign were observed in missense mutations (Table 1). Missense mutations generally disrupt the normal biological functions of a specific domain of the protein by changing the affinity of protein–protein interactions. Figure 3B shows the regions of missense mutations in the CASK protein found in patients, and the missense mutations associated with MICHPCH/microcephaly are confined to CaMK, PDZ, and SH3 domains, indicating that these domains play important roles in brain development, especially in the cerebellum.
MICPCH/microcephaly is one of the phenotypes of CASK-related disorders that has been extensively examined in animal models. The cerebellar size of cask heterozygous knockout female mice and hypomorphic male mice was shown to be smaller than that in wild-type mice [4,92,94,95]. Similarly, we reported that the knockout of neurexin, a main binding partner of CASK, from the cerebellar granule cells, induced cell death. The survival of cerebellar granule cells crucially depends on the presence of neurexin-1, -2, and -3 isoforms [96]. Intriguingly, in vitro studies demonstrated that the intracellular domain of neurexin, which includes the PDZ domain, was essential to the survival of cerebellar granule cells. The importance of the PDZ domain in cerebellar development is supported not only by genetic findings of missense mutations in the PDZ domain but also by our recent report using cerebellar granule cell culture. We demonstrated that the death of cerebellar granule cells from the conditional knockout of CASK can be rescued by the full length of CASK but not by the variants lacking the CaMK, PDZ, or SH3 domains of CASK, indicating that all three domains are essential for the survival of cerebellar granule cells in mice [95]. Moreover, one of the MICPCH-associated missense mutations (M519T), which could not bind to neurexin in vitro assay [63], failed to rescue the cell death of cerebellar granule cells from the knockout of the cask gene [95], indicating that CASK–neurexin binding would be important to cerebellar development. Although the molecular mechanism of cerebellar granule cell death is still unclear, one possible hypothesis is the neurexin-dependent release of brain-derived neurotrophic factor (BDNF) [96], which plays an essential role in cerebellar development in mice and primates [97,98,99].
In addition to the PDZ domain, other functional domains of the CASK protein are known to play important roles in binding CASK and neurexin. For instance, the CaMK domain of CASK does not include a known binding motif to neurexin but has been reported to bind to other proteins such as Liprin-α and Mint1 [25,37]. These molecules have been demonstrated to stabilize the binding between CASK and neurexin [100]. Thus, the CASK–neurexin–Liprin-α tripartite complex may be a molecular mechanism of the development of the cerebellum. Interestingly, a mutation in the CaMK domain, R106P, which was identified from a male patient of MICPCH [57], did not prevent the cell death of cerebellar granule cells [95]. The specific residue has been shown to contribute to interactions between CASK and Liprin-α and Mint1 [25], and an AlphaFold2-prediction of CaMK protein structure revealed that the mutation disrupted the interactions [95]. The contribution of CASK–neurexin interactions to cerebellar development is unclear, partially because limited information is available on the molecular structure between CASK and neurexins. More evidence on the molecular mechanism will help to understand that the interaction between CASK and neurexin plays an important role in the pathogenesis of MICPCH caused by CASK deficiency.
Another phenotype of CASK-related disorders is infantile epileptic syndrome, occurring in a smaller proportion of patients with CASK-related disorders (53.6% in males and 35.4% in females). Giacomini et al. reported a cohort of 34 patients with CASK-related disorders and reported that half of the patients suffered from epileptic syndromes. Among them, a main phenotype of epilepsy by CASK deficiency was late-onset drug-resistant spasms, some of which developed into developmental and epileptic encephalopathy [10]. It has also been reported that patients with CASK-related disorder can be affected by Ohtahara syndrome [6], West syndrome [9,62], absences epilepsy [5,11,14], Lennox-Gastaut syndrome [7], and myoclonic epilepsy [7,54]. Thus, epileptic phenotypes of CASK-related disorders are diverse among patients. Fifteen missense mutations have been identified in association with epileptic seizures, and the locations of these mutations do not converge on specific domains but diverge over all the functional domains of the CASK protein (Figure 3B). The wide distribution of missense mutations of CASK in the patients of epilepsy, which is also observed in the case of intellectual disability, indicates that epilepsy and intellectual disability may be due to multiple molecular mechanisms related to different functional domains of the CASK protein.
One of the CASK-partner molecules associated with infantile epilepsy is the NMDA receptor 2B subunit (GluN2B). Various mutations in the gene of GluN2B were found in patients suffering from infantile epilepsy [101,102]. The administration of NMDA has been used as a rodent model of infantile epilepsy [103,104,105]. The guanylate kinase domain at the C-terminal of CASK interacts with T-box transcription factor-1 (TBR1) and CASK regulates the gene expression of GluN2B in a TBR1-dependent manner [40,43]. We recently examined neuronal functional changes caused by the disruption of the CASK-TBR1-GluN2B pathway [92]. We first recorded cortical neurons of CASK heterozygous knockout mice and genotyped the recorded neurons using single-cell RT-PCR. We found that approximately 50% of neurons were CASK-positive, supporting a notion of the random distribution of CASK-expressing cells by XCI. Synaptic inputs to the CASK-expressing neurons in the heterozygous knockout mice were not different from those in the wild-type mice. On the other hand, the CASK-deficient neurons received more excitatory and fewer inhibitory synapses. The imbalance of excitatory and inhibitory synaptic inputs was due to the downregulation of GluN2B caused by the disruption of the CASK–TBR1 interaction. The increased excitability of neuronal circuits caused by the loss of CASK gene expression may explain the pathogenesis of epilepsy in patients with CASK-related disorders. The synaptic inputs to CASK-deficient neurons are stronger than those to CASK-expressing neurons, suggesting that CASK-deficient neuronal circuits are preferentially connected.
5. Unresolved Questions on the Pathophysiology of CASK-Related Disorders
Genetic studies of patients with genetic abnormalities in CASK and studies using cask knockout mice [106] are expected to help understand the pathogenesis of CASK-related disorders and to develop therapeutic strategies. However, to achieve this, some contradictions between human and animal-model studies must be resolved and understood in an integrated manner.
MICPCH syndrome is one of the pathogenic phenotypes of CASK-related disorders, for which there are differences between human phenotypes and mouse models. Magnetic resonance imaging (MRI) cerebellar morphology has been reported in approximately 50% of cases, with significantly smaller cerebellar volumes in both boys and girls, and some reports of cerebellar atrophy showed as much as 90% was abnormal [4]. In patients with CASK-related disorders, microcephaly has been shown to be progressive from the prenatal period [107]. On the other hand, with respect to studies in animal models, Najm et al. reported a reduction in cerebellar cortical thickness in cask-flox mice in which the expression of the cask gene decreased by about 20% (Supplementary Figure S5 in [4]). Mukherjee’s group has reported that the size of the cerebellum was 40% smaller in cask heterozygous knockout mice [79,92,94], which is still moderate compared with human cases. It has also been reported that the cerebellum size of cask heterozygous knockout mice is almost the same as that of the wild type during the first week after birth, after which the development of the cerebellum stagnates [95]. This cerebellar stagnation and atrophy may not be limited to early development; Patel et al. studied mice in which the CASK gene was deleted in most of the cells that make up the cerebellum [79]. In those mice, they reported a decrease in cerebellar size after 2 months of age, accompanied by the observation of cerebellar-dependent ataxia. These differences in the process of microcephaly and MICPCH between humans and mouse models are thought to be largely due to interspecies differences in neurodevelopment, and differences in the mutation patterns of the CASK gene. The lethality of the constitutive cask knockout has hindered the development of such studies. The introduction and analysis of these new methods are expected to provide answers to these questions.
No trend has been observed in pathogenic mutations of the CASK gene related to ID and epilepsy to date. Considering that each functional domain exerts a unique biological function by associating with different partner proteins, it is reasonable to assume that there are multiple molecular mechanisms underlying the ID or epilepsy in CASK-related disorders. Intellectual disability and epilepsy are known to be caused by genetic mutations in CASK partner proteins, including adhesion factors involved in synapse formation (e.g., Neurexin), presynaptic-release-related factors (voltage-gated calcium channels, SNARE-related molecules), and postsynaptic receptors (NMDA receptors) [108]. Interactions between CASK and the partner proteins must be examined to understand the mechanisms of CASK-associated intellectual disability and epilepsy and to develop new therapeutic strategies. Until recently, these protein–protein interactions were examined by biochemical assays targeting a few molecular mechanisms of interest. In addition to these classical approaches, the use of machine learning to predict higher-order protein structures, which has developed remarkably in recent years, is expected to dramatically improve our understanding of the protein–protein interactions underlying the pathogenesis of CASK-related disorders on a larger scale. To our knowledge, all CASK protein variants are speculated to be loss-of-function or hypomorphic variants. The improved prediction of protein structure and protein–protein interactions based on machine learning will aid in the discovery of unknown gain-of-function mutations that result in clinical phenotypes.
In addition to CASK-related disorders, several other X-linked neurological diseases, such as PCDH19-related disorders [109], have been reported in which most patients were female. These diseases exhibit a range of clinical symptoms that vary in severity among individuals. One possible explanation for this variability is the proportion of gene-deficient neurons in an individual’s brain, which may be correlated with the severity of symptoms (Figure 4A). As previously discussed, epilepsy phenotypes were observed more frequently in males with CASK-related disorders than in females. This sexual difference in epilepsy incidence may be due to the proportion of CASK-expressing cells in the brain. It has also been demonstrated that CASK-related disorders may comprise relatively separate neuronal circuits with different physiological characteristics in a genotype-dependent manner. The cellular interference hypothesis, which proposes that interference between these two types of genetically distinct neuronal circuits results in the disruption of neuronal function, has been proposed to explain the pathophysiology of PCDH19-related disorders. This concept of neurocircuit interference may provide new insights into the neurocircuit mechanism of female-restricted intellectual disability and epilepsy in CASK-related disorders, as well as other X-linked disorders (Figure 4B).
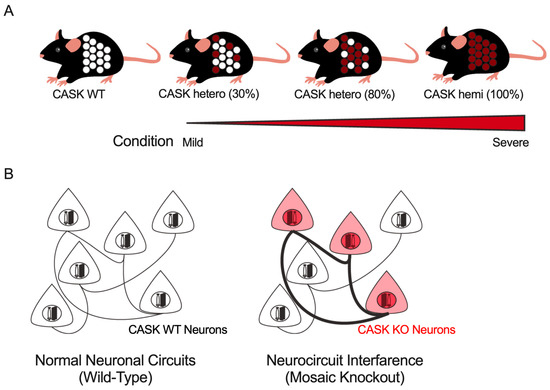
Figure 4.
Mesoscopic hypotheses underlying the pathophysiology of CASK-related disorders as well as other X-linked neurological disorders in female patients. (A) All the somatic cells in male patients express a pathogenic mutant of the CASK protein (the rightmost), except for somatic mosaic cases of males. Female patients consist of a mixture of CASK-expressing (white) and -deficient (red) cells and the proportion of the two genotypes of cells may determine the severity of CASK-related disorders. (B) CASK-deficient neurons (red) are connected more preferentially to each other, and synaptic connections between CASK-expressing neurons are weaker. The “neurocircuit interference”, a genotype-independent neural circuitry, may explain the pathogenesis of CASK-related disorders.
To validate these hypotheses, it is necessary to simultaneously examine the pathology of the patient and the XCI pattern in the brain, but it is technically almost impossible to directly examine the XCI pattern in the patient’s brain. To address this challenge, researchers are turning to alternative approaches, such as utilizing mouse models that enable the simultaneous analysis of phenotypes and the brain XCI patterns. Such models offer a valuable tool to verify the hypotheses and contribute to the development of methods for estimating brain XCI patterns.
6. Conclusions
In this review, we have discussed significant associations between CASK gene mutation patterns and the pathophysiology of CASK-related disorders. Additionally, we described the molecular interactions that are necessary for understanding the molecular mechanisms of CASK-related disorders. Although we mainly focus on MICPCH and ID/epilepsy in this review, CASK-related disorders also include a wide range of clinical manifestations, including ophthalmologic conditions such as nystagmus, sensorineural hearing loss, short stature, and structural abnormalities of the heart, which were not covered in this review. Therefore, it is essential to elucidate the genetic background of these conditions through the further follow-up of patients with CASK-related disorders. Collaboration with the recently organized groups of patients with CASK-related disorders is expected to provide information on the pathogenesis of CASK-related disorders and the mode of genetic variation contributing to its development. Based on this comprehensive clinical information, we expect that an analysis of interactions with CASK using machine learning to predict the protein conformation and the generation of mouse models carrying specific genetic mutations from patients using genome editing will contribute to the development of future treatments for CASK-related disorders.
Author Contributions
Writing—original draft preparation, T.M., M.Z. and K.T.; funding acquisition, T.M. and K.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by KAKENHI 21K07293 (T.M.) and 23H02575 (K.T.), Grant-in-Aid for Transformative Research Areas (A) 21H05685 (T.M.) and 23H04227 (K.T.); the Japan Epilepsy Research Foundation (T.M.), the Takeda Science Foundation (T.M. and K.T.), Naito Foundation (T.M.), Hokuto Foundation (T.M.), the Mochida Memorial Foundation for Medical and Pharmaceutical Research (T.M.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We are grateful to all the Tabuchi Lab members for helpful suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Cohen, A.R.; Woods, D.F.; Marfatia, S.M.; Walther, Z.; Chishti, A.H.; Anderson, J.M. Human CASK/LIN-2 binds syndecan-2 and protein 4.1 and localizes to the basolateral membrane of epithelial cells. J. Cell Biol. 1998, 142, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Dimitratos, S.D.; Stathakis, D.G.; Nelson, C.A.; Woods, D.F.; Bryant, P.J. The location of human CASK at Xp11.4 identifies this gene as a candidate for X-linked optic atrophy. Genomics 1998, 51, 308–309. [Google Scholar] [CrossRef] [PubMed]
- Froyen, G.; Van Esch, H.; Bauters, M.; Hollanders, K.; Frints, S.G.; Vermeesch, J.R.; Devriendt, K.; Fryns, J.P.; Marynen, P. Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: Important role for increased gene dosage of XLMR genes. Hum. Mutat. 2007, 28, 1034–1042. [Google Scholar] [CrossRef]
- Najm, J.; Horn, D.; Wimplinger, I.; Golden, J.A.; Chizhikov, V.V.; Sudi, J.; Christian, S.L.; Ullmann, R.; Kuechler, A.; Haas, C.A.; et al. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet. 2008, 40, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Burglen, L.; Chantot-Bastaraud, S.; Garel, C.; Milh, M.; Touraine, R.; Zanni, G.; Petit, F.; Afenjar, A.; Goizet, C.; Barresi, S.; et al. Spectrum of pontocerebellar hypoplasia in 13 girls and boys with CASK mutations: Confirmation of a recognizable phenotype and first description of a male mosaic patient. Orphanet. J. Rare Dis. 2012, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Kato, M.; Osaka, H.; Moriyama, N.; Horita, H.; Nishiyama, K.; Yoneda, Y.; Kondo, Y.; Tsurusaki, Y.; Doi, H.; et al. CASK aberrations in male patients with Ohtahara syndrome and cerebellar hypoplasia. Epilepsia 2012, 53, 1441–1449. [Google Scholar] [CrossRef]
- Takanashi, J.; Okamoto, N.; Yamamoto, Y.; Hayashi, S.; Arai, H.; Takahashi, Y.; Maruyama, K.; Mizuno, S.; Shimakawa, S.; Ono, H.; et al. Clinical and radiological features of Japanese patients with a severe phenotype due to CASK mutations. Am. J. Med. Genet. A 2012, 158A, 3112–3118. [Google Scholar] [CrossRef]
- Nakamura, K.; Nishiyama, K.; Kodera, H.; Nakashima, M.; Tsurusaki, Y.; Miyake, N.; Matsumoto, N.; Saitsu, H.; Jinnou, H.; Ohki, S.; et al. A de novo CASK mutation in pontocerebellar hypoplasia type 3 with early myoclonic epilepsy and tetralogy of Fallot. Brain Dev. 2014, 36, 272–273. [Google Scholar] [CrossRef]
- Nakajiri, T.; Kobayashi, K.; Okamoto, N.; Oka, M.; Miya, F.; Kosaki, K.; Yoshinaga, H. Late-onset epileptic spasms in a female patient with a CASK mutation. Brain Dev. 2015, 37, 919–923. [Google Scholar] [CrossRef]
- Giacomini, T.; Nuovo, S.; Zanni, G.; Mancardi, M.M.; Cusmai, R.; Pepi, C.; Bertini, E.; Valente, E.M.; Battini, R.; Ferrari, A.; et al. CASK related disorder: Epilepsy and developmental outcome. Eur. J. Paediatr. Neurol. 2021, 31, 61–69. [Google Scholar] [CrossRef]
- Moog, U.; Kutsche, K.; Kortüm, F.; Chilian, B.; Bierhals, T.; Apeshiotis, N.; Balg, S.; Chassaing, N.; Coubes, C.; Das, S.; et al. Phenotypic spectrum associated with CASK loss-of-function mutations. J. Med. Genet. 2011, 48, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Piluso, G.; D’Amico, F.; Saccone, V.; Bismuto, E.; Rotundo, I.L.; Di Domenico, M.; Aurino, S.; Schwartz, C.E.; Neri, G.; Nigro, V. A missense mutation in CASK causes FG syndrome in an Italian family. Am. J. Hum. Genet. 2009, 84, 162–177. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dunn, P.; Prigatano, G.P.; Szelinger, S.; Roth, J.; Siniard, A.L.; Claasen, A.M.; Richholt, R.F.; De Both, M.; Corneveaux, J.J.; Moskowitz, A.M.; et al. A de novo splice site mutation in CASK causes FG syndrome-4 and congenital nystagmus. Am. J. Med. Genet. A 2017, 173, 611–617. [Google Scholar] [CrossRef]
- Hackett, A.; Tarpey, P.S.; Licata, A.; Cox, J.; Whibley, A.; Boyle, J.; Rogers, C.; Grigg, J.; Partington, M.; Stevenson, R.E.; et al. CASK mutations are frequent in males and cause X-linked nystagmus and variable XLMR phenotypes. Eur. J. Hum. Genet. 2010, 18, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.; Mastropasqua, F.; Reising, J.P.; Maier, S.; Ho, M.L.; Rabkina, I.; Li, D.; Neufeld, J.; Ballenberger, L.; Myers, L.; et al. Presynaptic dysfunction in CASK-related neurodevelopmental disorders. Transl. Psychiatry 2020, 10, 312. [Google Scholar] [CrossRef]
- Mustroph, J.; Sag, C.M.; Bähr, F.; Schmidtmann, A.L.; Gupta, S.N.; Dietz, A.; Islam, M.M.T.; Lücht, C.; Beuthner, B.E.; Pabel, S.; et al. Loss of CASK Accelerates Heart Failure Development. Circ. Res. 2021, 128, 1139–1155. [Google Scholar] [CrossRef]
- Wei, J.L.; Fu, Z.X.; Fang, M.; Zhou, Q.Y.; Zhao, Q.N.; Guo, J.B.; Lu, W.D.; Wang, H. High expression of CASK correlates with progression and poor prognosis of colorectal cancer. Tumor Biol. 2014, 35, 9185–9194. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, G.; Yin, C.; Jin, W.; Zhang, G. Down-regulation of miR-203 induced by Helicobacter pylori infection promotes the proliferation and invasion of gastric cancer by targeting CASK. Oncotarget 2014, 5, 11631–11640. [Google Scholar] [CrossRef]
- Ding, B.; Bao, C.; Jin, L.; Xu, L.; Fan, W.; Lou, W. CASK Silence Overcomes Sorafenib Resistance of Hepatocellular Carcinoma through Activating Apoptosis and Autophagic Cell Death. Front. Oncol. 2021, 11, 681683. [Google Scholar] [CrossRef]
- Qu, J.; Zhou, Y.; Li, Y.; Yu, J.; Wang, W. CASK regulates Notch pathway and functions as a tumor promoter in pancreatic cancer. Arch. Biochem. Biophys. 2021, 701, 108789. [Google Scholar] [CrossRef]
- Hata, Y.; Butz, S.; Südhof, T.C. CASK: A novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J. Neurosci. 1996, 16, 2488–2494. [Google Scholar] [CrossRef] [PubMed]
- Baines, A.J. Caenorhabditis elegans LIN-2A and mammalian neuronal CASK are prototypical members of a subfamily of MAGUKs (membrane-associated guanylate kinases) characterized by a common kinase-like domain and a guanylate kinase domain predicted to bind ATP. Biochem. J. 1996, 320 Pt 2, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Dimitratos, S.D.; Woods, D.F.; Bryant, P.J. Camguk, Lin-2, and CASK: Novel membrane-associated guanylate kinase homologs that also contain CaM kinase domains. Mech. Dev. 1997, 63, 127–130. [Google Scholar] [CrossRef]
- Mukherjee, K.; Sharma, M.; Urlaub, H.; Bourenkov, G.P.; Jahn, R.; Südhof, T.C.; Wahl, M.C. CASK Functions as a Mg2+-independent neurexin kinase. Cell 2008, 133, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Zheng, S.; Spangler, S.A.; Yu, C.; Hoogenraad, C.C.; Zhang, M. Liprin-mediated large signaling complex organization revealed by the liprin-α/CASK and liprin-α/liprin-β complex structures. Mol. Cell 2011, 43, 586–598. [Google Scholar] [CrossRef]
- Spangler, S.A.; Schmitz, S.K.; Kevenaar, J.T.; de Graaff, E.; de Wit, H.; Demmers, J.; Toonen, R.F.; Hoogenraad, C.C. Liprin-α2 promotes the presynaptic recruitment and turnover of RIM1/CASK to facilitate synaptic transmission. J. Cell Biol. 2013, 201, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Leonoudakis, D.; Conti, L.R.; Radeke, C.M.; McGuire, L.M.; Vandenberg, C.A. A multiprotein trafficking complex composed of SAP97, CASK, Veli, and Mint1 is associated with inward rectifier Kir2 potassium channels. J. Biol. Chem. 2004, 279, 19051–19063. [Google Scholar] [CrossRef]
- Hong, C.J.; Hsueh, Y.P. CASK associates with glutamate receptor interacting protein and signaling molecules. Biochem. Biophys. Res. Commun. 2006, 351, 771–776. [Google Scholar] [CrossRef]
- Stafford, R.L.; Ear, J.; Knight, M.J.; Bowie, J.U. The molecular basis of the Caskin1 and Mint1 interaction with CASK. J. Mol. Biol. 2011, 412, 3–13. [Google Scholar] [CrossRef]
- Lee, S.; Fan, S.; Makarova, O.; Straight, S.; Margolis, B. A novel and conserved protein-protein interaction domain of mammalian Lin-2/CASK binds and recruits SAP97 to the lateral surface of epithelia. Mol. Cell. Biol. 2002, 22, 1778–1791. [Google Scholar] [CrossRef]
- Jeyifous, O.; Waites, C.L.; Specht, C.G.; Fujisawa, S.; Schubert, M.; Lin, E.I.; Marshall, J.; Aoki, C.; de Silva, T.; Montgomery, J.M.; et al. SAP97 and CASK mediate sorting of NMDA receptors through a previously unknown secretory pathway. Nat. Neurosci. 2009, 12, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Olsen, O.; Liu, H.; Wade, J.B.; Merot, J.; Welling, P.A. Basolateral membrane expression of the Kir 2.3 channel is coordinated by PDZ interaction with Lin-7/CASK complex. Am. J. Physiol. Cell Physiol. 2002, 282, C183–C195. [Google Scholar] [CrossRef] [PubMed]
- Alewine, C.; Kim, B.Y.; Hegde, V.; Welling, P.A. Lin-7 targets the Kir 2.3 channel on the basolateral membrane via a L27 domain interaction with CASK. Am. J. Physiol. Cell Physiol. 2007, 293, C1733–C1741. [Google Scholar] [CrossRef]
- Hsueh, Y.P.; Yang, F.C.; Kharazia, V.; Naisbitt, S.; Cohen, A.R.; Weinberg, R.J.; Sheng, M. Direct interaction of CASK/LIN-2 and syndecan heparan sulfate proteoglycan and their overlapping distribution in neuronal synapses. J. Cell Biol. 1998, 142, 139–151. [Google Scholar] [CrossRef]
- Hsueh, Y.P.; Sheng, M. Regulated expression and subcellular localization of syndecan heparan sulfate proteoglycans and the syndecan-binding protein CASK/LIN-2 during rat brain development. J. Neurosci. 1999, 19, 7415–7425. [Google Scholar] [CrossRef] [PubMed]
- Gomez, A.M.; Traunmüller, L.; Scheiffele, P. Neurexins: Molecular codes for shaping neuronal synapses. Nat. Rev. Neurosci. 2021, 22, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Maximov, A.; Südhof, T.C.; Bezprozvanny, I. Association of neuronal calcium channels with modular adaptor proteins. J. Biol. Chem. 1999, 274, 24453–24456. [Google Scholar] [CrossRef]
- Maximov, A.; Bezprozvanny, I. Synaptic targeting of N-type calcium channels in hippocampal neurons. J. Neurosci. 2002, 22, 6939–6952. [Google Scholar] [CrossRef]
- Hsueh, Y.P.; Wang, T.F.; Yang, F.C.; Sheng, M. Nuclear translocation and transcription regulation by the membrane-associated guanylate kinase CASK/LIN-2. Nature 2000, 404, 298–302. [Google Scholar] [CrossRef]
- Wang, G.S.; Hong, C.J.; Yen, T.Y.; Huang, H.Y.; Ou, Y.; Huang, T.N.; Jung, W.G.; Kuo, T.Y.; Sheng, M.; Wang, T.F.; et al. Transcriptional modification by a CASK-interacting nucleosome assembly protein. Neuron 2004, 42, 113–128. [Google Scholar] [CrossRef]
- Lin, C.W.; Huang, T.N.; Wang, G.S.; Kuo, T.Y.; Yen, T.Y.; Hsueh, Y.P. Neural activity- and development-dependent expression and distribution of CASK interacting nucleosome assembly protein in mouse brain. J. Comp. Neurol. 2006, 494, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lu, J.; Yang, C.; Wang, X.; Cheng, L.; Hu, G.; Sun, Y.; Zhang, X.; Wu, M.; Liu, Z. CASK and its target gene Reelin were co-upregulated in human esophageal carcinoma. Cancer Lett. 2002, 179, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.N.; Hsueh, Y.P. CASK point mutation regulates protein-protein interactions and NR2b promoter activity. Biochem. Biophys. Res. Commun. 2009, 382, 219–222. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Pan, Y.E.; Tibbe, D.; Harms, F.L.; Reißner, C.; Becker, K.; Dingmann, B.; Mirzaa, G.; Kattentidt-Mouravieva, A.A.; Shoukier, M.; Aggarwal, S.; et al. Missense mutations in CASK, coding for the calcium-/calmodulin-dependent serine protein kinase, interfere with neurexin binding and neurexin-induced oligomerization. J. Neurochem. 2021, 157, 1331–1350. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Hayashi, S.; Mizuno, S.; Migita, O.; Okuyama, T.; Makita, Y.; Hata, A.; Imoto, I.; Inazawa, J. The CASK gene harbored in a deletion detected by array-CGH as a potential candidate for a gene causative of X-linked dominant mental retardation. Am. J. Med. Genet. A 2008, 146A, 2145–2151. [Google Scholar] [CrossRef]
- Tarpey, P.S.; Smith, R.; Pleasance, E.; Whibley, A.; Edkins, S.; Hardy, C.; O’Meara, S.; Latimer, C.; Dicks, E.; Menzies, A.; et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009, 41, 535–543. [Google Scholar] [CrossRef]
- Hayashi, S.; Okamoto, N.; Chinen, Y.; Takanashi, J.; Makita, Y.; Hata, A.; Imoto, I.; Inazawa, J. Novel intragenic duplications and mutations of CASK in patients with mental retardation and microcephaly with pontine and cerebellar hypoplasia (MICPCH). Hum. Genet. 2012, 131, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Valayannopoulos, V.; Michot, C.; Rodriguez, D.; Hubert, L.; Saillour, Y.; Labrune, P.; de Laveaucoupet, J.; Brunelle, F.; Amiel, J.; Lyonnet, S.; et al. Mutations of TSEN and CASK genes are prevalent in pontocerebellar hypoplasias type 2 and 4. Brain 2012, 135, e199; author reply e200. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.L.; Lachance, M.; Hamdan, F.F.; Carmant, L.; Lortie, A.; Diadori, P.; Major, P.; Meijer, I.A.; Lemyre, E.; Cossette, P.; et al. The genetic landscape of infantile spasms. Hum. Mol. Genet. 2014, 23, 4846–4858. [Google Scholar] [CrossRef] [PubMed]
- Moog, U.; Bierhals, T.; Brand, K.; Bautsch, J.; Biskup, S.; Brune, T.; Denecke, J.; de Die-Smulders, C.E.; Evers, C.; Hempel, M.; et al. Phenotypic and molecular insights into CASK-related disorders in males. Orphanet. J. Rare Dis. 2015, 10, 44. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rump, P.; Jazayeri, O.; van Dijk-Bos, K.K.; Johansson, L.F.; van Essen, A.J.; Verheij, J.B.; Veenstra-Knol, H.E.; Redeker, E.J.; Mannens, M.M.; Swertz, M.A.; et al. Whole-exome sequencing is a powerful approach for establishing the etiological diagnosis in patients with intellectual disability and microcephaly. BMC Med. Genom. 2016, 9, 7. [Google Scholar] [CrossRef]
- Rivas, L.; Blanco, Ó.; Torreira, C.; Repáraz, A.; Melcón, C.; Amado, A. Pontocerebellar hypoplasia secondary to CASK gene deletion: Case report. Rev. Chil. Pediatr. 2017, 88, 529–533. [Google Scholar] [CrossRef]
- Hayashi, S.; Uehara, D.T.; Tanimoto, K.; Mizuno, S.; Chinen, Y.; Fukumura, S.; Takanashi, J.I.; Osaka, H.; Okamoto, N.; Inazawa, J. Comprehensive investigation of CASK mutations and other genetic etiologies in 41 patients with intellectual disability and microcephaly with pontine and cerebellar hypoplasia (MICPCH). PLoS ONE 2017, 12, e0181791. [Google Scholar] [CrossRef]
- Popp, B.; Ekici, A.B.; Thiel, C.T.; Hoyer, J.; Wiesener, A.; Kraus, C.; Reis, A.; Zweier, C. Exome Pool-Seq in neurodevelopmental disorders. Eur. J. Hum. Genet. 2017, 25, 1364–1376. [Google Scholar] [CrossRef]
- DeLuca, S.C.; Wallace, D.A.; Trucks, M.R.; Mukherjee, K. A clinical series using intensive neurorehabilitation to promote functional motor and cognitive skills in three girls with CASK mutation. BMC Res. Notes 2017, 10, 743. [Google Scholar] [CrossRef]
- Seto, T.; Hamazaki, T.; Nishigaki, S.; Kudo, S.; Shintaku, H.; Ondo, Y.; Shimojima, K.; Yamamoto, T. A novel CASK mutation identified in siblings exhibiting developmental disorders with/without microcephaly. Intractable Rare Dis. Res. 2017, 6, 177–182. [Google Scholar] [CrossRef]
- Muthusamy, B.; Selvan, L.D.N.; Nguyen, T.T.; Manoj, J.; Stawiski, E.W.; Jaiswal, B.S.; Wang, W.; Raja, R.; Ramprasad, V.L.; Gupta, R.; et al. Next-Generation Sequencing Reveals Novel Mutations in X-linked Intellectual Disability. OMICS 2017, 21, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Bozarth, X.; Foss, K.; Mefford, H.C. A de novo in-frame deletion of CASK gene causes early onset infantile spasms and supratentorial cerebral malformation in a female patient. Am. J. Med. Genet. A 2018, 176, 2425–2429. [Google Scholar] [CrossRef]
- LaConte, L.E.W.; Chavan, V.; Elias, A.F.; Hudson, C.; Schwanke, C.; Styren, K.; Shoof, J.; Kok, F.; Srivastava, S.; Mukherjee, K. Two microcephaly-associated novel missense mutations in CASK specifically disrupt the CASK-neurexin interaction. Hum. Genet. 2018, 137, 231–246. [Google Scholar] [CrossRef] [PubMed]
- LaConte, L.E.W.; Chavan, V.; DeLuca, S.; Rubin, K.; Malc, J.; Berry, S.; Gail Summers, C.; Mukherjee, K. An N-terminal heterozygous missense CASK mutation is associated with microcephaly and bilateral retinal dystrophy plus optic nerve atrophy. Am. J. Med. Genet. A 2019, 179, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Cristofoli, F.; Devriendt, K.; Davis, E.E.; Van Esch, H.; Vermeesch, J.R. Novel CASK mutations in cases with syndromic microcephaly. Hum. Mutat. 2018, 39, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Hauer, N.N.; Popp, B.; Schoeller, E.; Schuhmann, S.; Heath, K.E.; Hisado-Oliva, A.; Klinger, P.; Kraus, C.; Trautmann, U.; Zenker, M.; et al. Clinical relevance of systematic phenotyping and exome sequencing in patients with short stature. Genet. Med. 2018, 20, 630–638. [Google Scholar] [CrossRef]
- Murakami, H.; Kimura, Y.; Enomoto, Y.; Tsurusaki, Y.; Akahira-Azuma, M.; Kuroda, Y.; Tsuji, M.; Goto, T.; Kurosawa, K. Discordant phenotype caused by CASK mutation in siblings with NF1. Hum. Genome Var. 2019, 6, 20. [Google Scholar] [CrossRef]
- Zhang, K.; Yuan, Q.; Xie, J.; Yuan, L.; Wang, Y. PPAR-γ activation increases insulin secretion independent of CASK in INS-1 cells. Acta Biochim. Biophys. Sin. 2019, 51, 715–722. [Google Scholar] [CrossRef]
- Aspromonte, M.C.; Bellini, M.; Gasparini, A.; Carraro, M.; Bettella, E.; Polli, R.; Cesca, F.; Bigoni, S.; Boni, S.; Carlet, O.; et al. Characterization of intellectual disability and autism comorbidity through gene panel sequencing. Hum. Mutat. 2019, 40, 1346–1363. [Google Scholar] [CrossRef]
- Rochtus, A.; Olson, H.E.; Smith, L.; Keith, L.G.; El Achkar, C.; Taylor, A.; Mahida, S.; Park, M.; Kelly, M.; Shain, C.; et al. Genetic diagnoses in epilepsy: The impact of dynamic exome analysis in a pediatric cohort. Epilepsia 2020, 61, 249–258. [Google Scholar] [CrossRef]
- Ibarluzea, N.; Hoz, A.B.; Villate, O.; Llano, I.; Ocio, I.; Martí, I.; Guitart, M.; Gabau, E.; Andrade, F.; Gener, B.; et al. Targeted Next-Generation Sequencing in Patients with Suggestive X-Linked Intellectual Disability. Genes 2020, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.X.; Ma, H.X.; Zhang, Y.X.; Chen, Z.H.; Zhai, Q.X. Whole-Exome Sequencing for Identifying Genetic Causes of Intellectual Developmental Disorders. Int. J. Gen. Med. 2021, 14, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Dubbs, H.; Ortiz-Gonzalez, X.; Marsh, E.D. Pathogenic variants in CASK: Expanding the genotype-phenotype correlations. Am. J. Med. Genet. A 2022, 188, 2617–2626. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Zhang, Y.; Yang, W.; Yang, L.; Wang, R.; Xu, M.; Sun, L.; Zhang, B.; Cui, X. Case report: A novel CASK mutation in a Chinese female child with microcephaly with pontine and cerebellar hypoplasia. Front. Genet. 2022, 13, 856636. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Jia, P.; Yao, Y.; Zhu, F. Case Report: Identification of a novel CASK missense variant in a Chinese family with MICPCH. Front. Genet. 2022, 13, 933785. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Jiang, C.; Li, J.; Zhang, G.; Shen, Y.; Wang, J. A novel missense variant in the CASK gene causes intellectual developmental disorder and microcephaly with pontine and cerebellar hypoplasia. BMC Med. Genom. 2022, 15, 127. [Google Scholar] [CrossRef]
- Zhang, Y.; Nie, Y.; Mu, Y.; Zheng, J.; Xu, X.; Zhang, F.; Shu, J.; Liu, Y. A de novo variant in CASK gene causing intellectual disability and brain hypoplasia: A case report and literature review. Ital. J. Pediatr. 2022, 48, 73. [Google Scholar] [CrossRef]
- Yang, K.; Lin, L.; Yuan, F.; Li, X.; Liu, Z.; Lan, X.; Wang, Y.; Ren, Y.; Li, J.; Chen, Y. Two heterozygous mutations in the calcium/calmodulin-dependent serine protein kinase gene (CASK) in cases with developmental disorders. Mol. Genet. Genom. Med. 2022, 10, e2065. [Google Scholar] [CrossRef]
- Patel, P.A.; Hegert, J.V.; Cristian, I.; Kerr, A.; LaConte, L.E.W.; Fox, M.A.; Srivastava, S.; Mukherjee, K. Complete loss of the X-linked gene CASK causes severe cerebellar degeneration. J. Med. Genet. 2022, 59, 1044–1057. [Google Scholar] [CrossRef]
- Lai, D.; Gade, M.; Yang, E.; Koh, H.Y.; Lu, J.; Walley, N.M.; Buckley, A.F.; Sands, T.T.; Akman, C.I.; Mikati, M.A.; et al. Somatic variants in diverse genes leads to a spectrum of focal cortical malformations. Brain 2022, 145, 2704–2720. [Google Scholar] [CrossRef]
- Tibbe, D.; Ferle, P.; Krisp, C.; Nampoothiri, S.; Mirzaa, G.; Assaf, M.; Parikh, S.; Kutsche, K.; Kreienkamp, H.J. Regulation of Liprin-α phase separation by CASK is disrupted by a mutation in its CaM kinase domain. Life Sci. Alliance 2022, 5, e202201512. [Google Scholar] [CrossRef] [PubMed]
- Abe-Hatano, C.; Yokoi, T.; Ida, K.; Kurosawa, K. Mosaicism of a Truncating Variant of CASK Causes Congenital Heart Disease and Neurodevelopmental Disorder. Mol. Syndromol. 2023, 13, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Long, S.; Zhou, H.; Li, S.; Wang, T.; Ma, Y.; Li, C.; Zhou, Y.; Zhou, S.; Wu, B.; Wang, Y. The Clinical and Genetic Features of Co-occurring Epilepsy and Autism Spectrum Disorder in Chinese Children. Front. Neurol. 2019, 10, 505. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [CrossRef]
- Tabuchi, K.; Blundell, J.; Etherton, M.R.; Hammer, R.E.; Liu, X.; Powell, C.M.; Südhof, T.C. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 2007, 318, 71–76. [Google Scholar] [CrossRef]
- Jamain, S.; Quach, H.; Betancur, C.; Råstam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef]
- Hansen, J.; Snow, C.; Tuttle, E.; Ghoneim, D.H.; Yang, C.S.; Spencer, A.; Gunter, S.A.; Smyser, C.D.; Gurnett, C.A.; Shinawi, M.; et al. De novo mutations in SIK1 cause a spectrum of developmental epilepsies. Am. J. Hum. Genet. 2015, 96, 682–690. [Google Scholar] [CrossRef]
- Badawi, M.; Mori, T.; Kurihara, T.; Yoshizawa, T.; Nohara, K.; Kouyama-Suzuki, E.; Yanagawa, T.; Shirai, Y.; Tabuchi, K. Risperidone Mitigates Enhanced Excitatory Neuronal Function and Repetitive Behavior Caused by an ASD-Associated Mutation of SIK1. Front. Mol. Neurosci. 2021, 14, 706494. [Google Scholar] [CrossRef]
- Borsani, G.; Tonlorenzi, R.; Simmler, M.C.; Dandolo, L.; Arnaud, D.; Capra, V.; Grompe, M.; Pizzuti, A.; Muzny, D.; Lawrence, C.; et al. Characterization of a murine gene expressed from the inactive X chromosome. Nature 1991, 351, 325–329. [Google Scholar] [CrossRef]
- Huynh, K.D.; Lee, J.T. X-chromosome inactivation: A hypothesis linking ontogeny and phylogeny. Nat. Rev. Genet. 2005, 6, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Kasem, E.A.; Suzuki-Kouyama, E.; Cao, X.; Li, X.; Kurihara, T.; Uemura, T.; Yanagawa, T.; Tabuchi, K. Deficiency of calcium/calmodulin-dependent serine protein kinase disrupts the excitatory-inhibitory balance of synapses by down-regulating GluN2B. Mol. Psychiatry 2019, 24, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; McMillan, R.; Willis, J.; Clark, H.; Chavan, V.; Liang, C.; Zhang, H.; Hulver, M.; Mukherjee, K. X-linked intellectual disability gene CASK regulates postnatal brain growth in a non-cell autonomous manner. Acta Neuropathol. Commun. 2016, 4, 30. [Google Scholar] [CrossRef]
- Guo, Q.; Kouyama-Suzuki, E.; Shirai, Y.; Cao, X.; Yanagawa, T.; Mori, T.; Tabuchi, K. Structural Analysis Implicates CASK-Liprin-α2 Interaction in Cerebellar Granular Cell Death in MICPCH Syndrome. Cells 2023, 12, 1177. [Google Scholar] [CrossRef]
- Uemura, T.; Suzuki-Kouyama, E.; Kawase, S.; Kurihara, T.; Yasumura, M.; Yoshida, T.; Fukai, S.; Yamazaki, M.; Fei, P.; Abe, M.; et al. Neurexins play a crucial role in cerebellar granule cell survival by organizing autocrine machinery for neurotrophins. Cell Rep. 2022, 39, 110624. [Google Scholar] [CrossRef]
- Mori, T.; Shimizu, K.; Hayashi, M. Differential expression patterns of TrkB ligands in the macaque monkey brain. Neuroreport 2004, 15, 2507–2511. [Google Scholar] [CrossRef]
- Schwartz, P.M.; Borghesani, P.R.; Levy, R.L.; Pomeroy, S.L.; Segal, R.A. Abnormal cerebellar development and foliation in BDNF−/− mice reveals a role for neurotrophins in CNS patterning. Neuron 1997, 19, 269–281. [Google Scholar] [CrossRef]
- Borghesani, P.R.; Peyrin, J.M.; Klein, R.; Rubin, J.; Carter, A.R.; Schwartz, P.M.; Luster, A.; Corfas, G.; Segal, R.A. BDNF stimulates migration of cerebellar granule cells. Development 2002, 129, 1435–1442. [Google Scholar] [CrossRef]
- LaConte, L.E.; Chavan, V.; Liang, C.; Willis, J.; Schönhense, E.M.; Schoch, S.; Mukherjee, K. CASK stabilizes neurexin and links it to liprin-α in a neuronal activity-dependent manner. Cell. Mol. Life Sci. 2016, 73, 3599–3621. [Google Scholar] [CrossRef]
- Lemke, J.R.; Hendrickx, R.; Geider, K.; Laube, B.; Schwake, M.; Harvey, R.J.; James, V.M.; Pepler, A.; Steiner, I.; Hörtnagel, K.; et al. GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann. Neurol. 2014, 75, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Platzer, K.; Yuan, H.; Schütz, H.; Winschel, A.; Chen, W.; Hu, C.; Kusumoto, H.; Heyne, H.O.; Helbig, K.L.; Tang, S.; et al. encephalopathy: Novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J. Med. Genet. 2017, 54, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.Y.; Yang, X.F.; Tomonoh, Y.; Hu, L.Y.; Ju, J.; Hirose, S.; Zou, L.P. Development of a mouse model of infantile spasms induced by N-methyl-D-aspartate. Epilepsy Res. 2015, 118, 29–33. [Google Scholar] [CrossRef]
- Pang, B.; Mori, T.; Badawi, M.; Zhou, M.; Guo, Q.; Suzuki-Kouyama, E.; Yanagawa, T.; Shirai, Y.; Tabuchi, K. An Epilepsy-Associated Mutation of Salt-Inducible Kinase 1 Increases the Susceptibility to Epileptic Seizures and Interferes with Adrenocorticotropic Hormone Therapy for Infantile Spasms in Mice. Int. J. Mol. Sci. 2022, 23, 7927. [Google Scholar] [CrossRef] [PubMed]
- Velísek, L.; Jehle, K.; Asche, S.; Velísková, J. Model of infantile spasms induced by N-methyl-D-aspartic acid in prenatally impaired brain. Ann. Neurol. 2007, 61, 109–119. [Google Scholar] [CrossRef]
- Atasoy, D.; Schoch, S.; Ho, A.; Nadasy, K.A.; Liu, X.; Zhang, W.; Mukherjee, K.; Nosyreva, E.D.; Fernandez-Chacon, R.; Missler, M.; et al. Deletion of CASK in mice is lethal and impairs synaptic function. Proc. Natl. Acad. Sci. USA 2007, 104, 2525–2530. [Google Scholar] [CrossRef] [PubMed]
- Gafner, M.; Boltshauser, E.; D’Abrusco, F.; Battini, R.; Romaniello, R.; D’Arrigo, S.; Zanni, G.; Leibovitz, Z.; Yosovich, K.; Lerman-Sagie, T.; et al. Expanding the natural history of CASK-related disorders to the prenatal period. Dev. Med. Child. Neurol. 2023, 65, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y.; Bear, M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, a009886. [Google Scholar] [CrossRef]
- Depienne, C.; Bouteiller, D.; Keren, B.; Cheuret, E.; Poirier, K.; Trouillard, O.; Benyahia, B.; Quelin, C.; Carpentier, W.; Julia, S.; et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet. 2009, 5, e1000381. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).