
Diverse Clinical Phenotypes of CASK-Related Disorders and Multiple Functional Domains of CASK Protein



Abstract
:1. CASK-Related Disorders, X-Linked Neurological Disorders
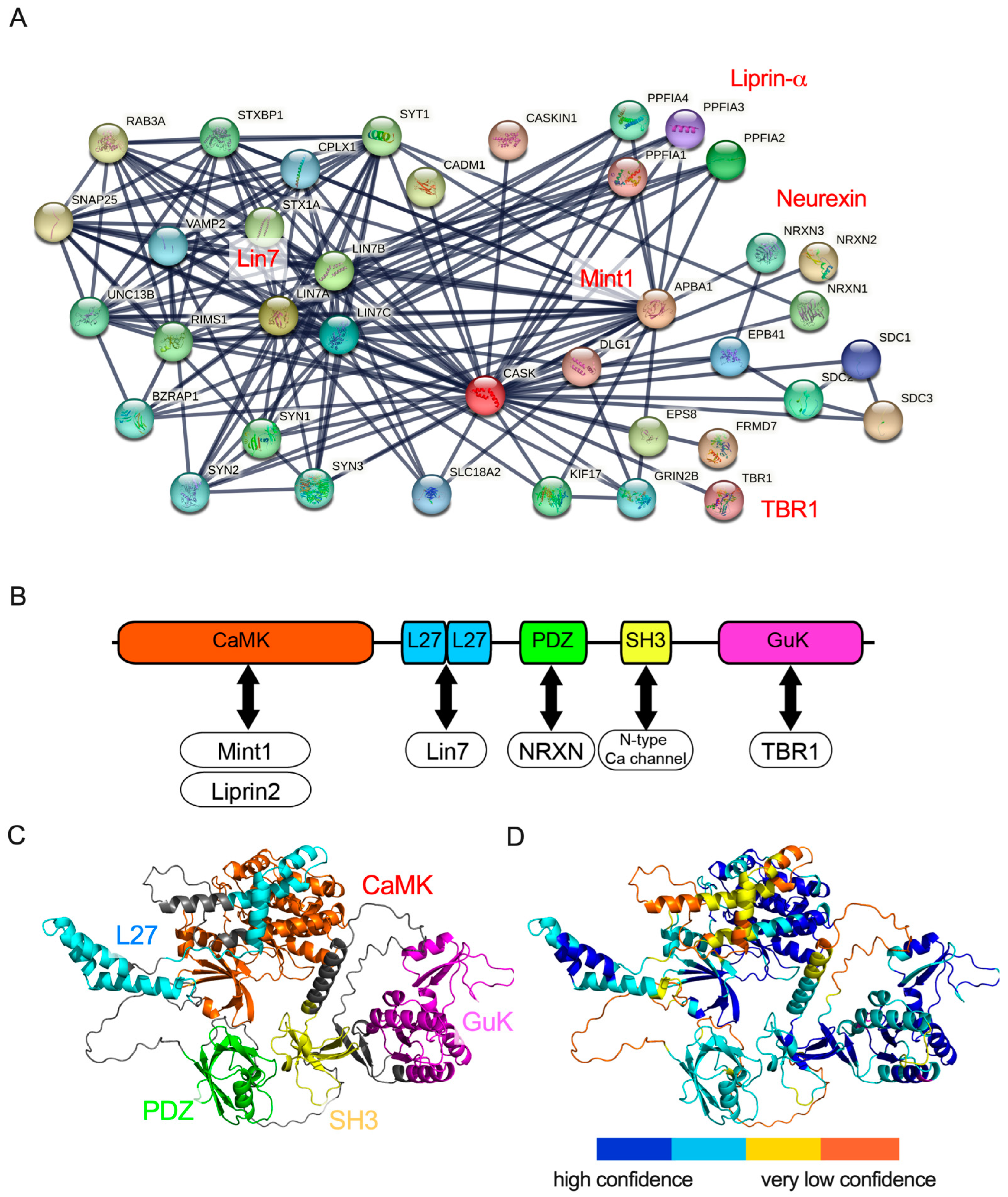
2. Protein Structure and Protein Interaction of CASK
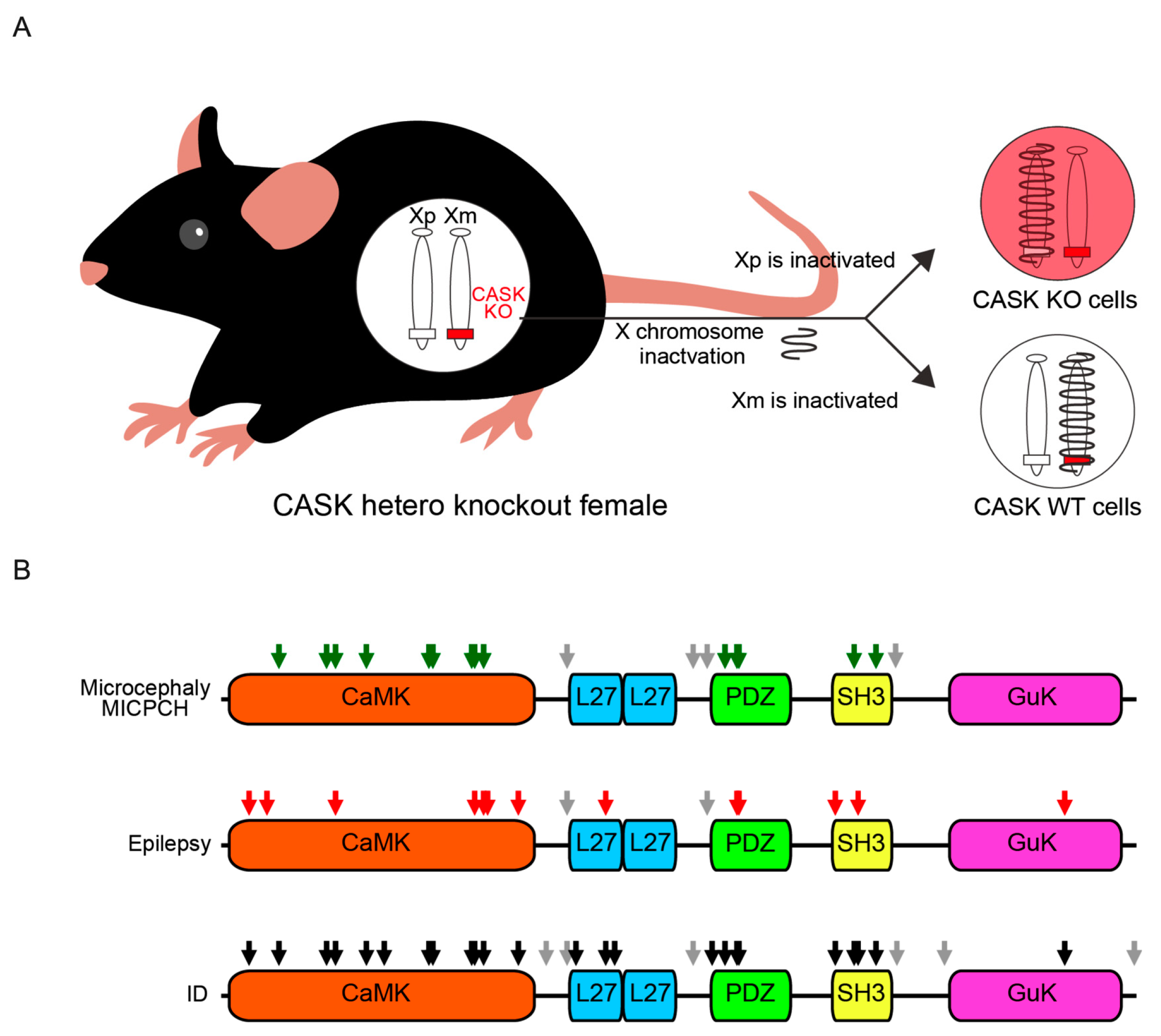
3. Genetics of CASK-Related Disorders
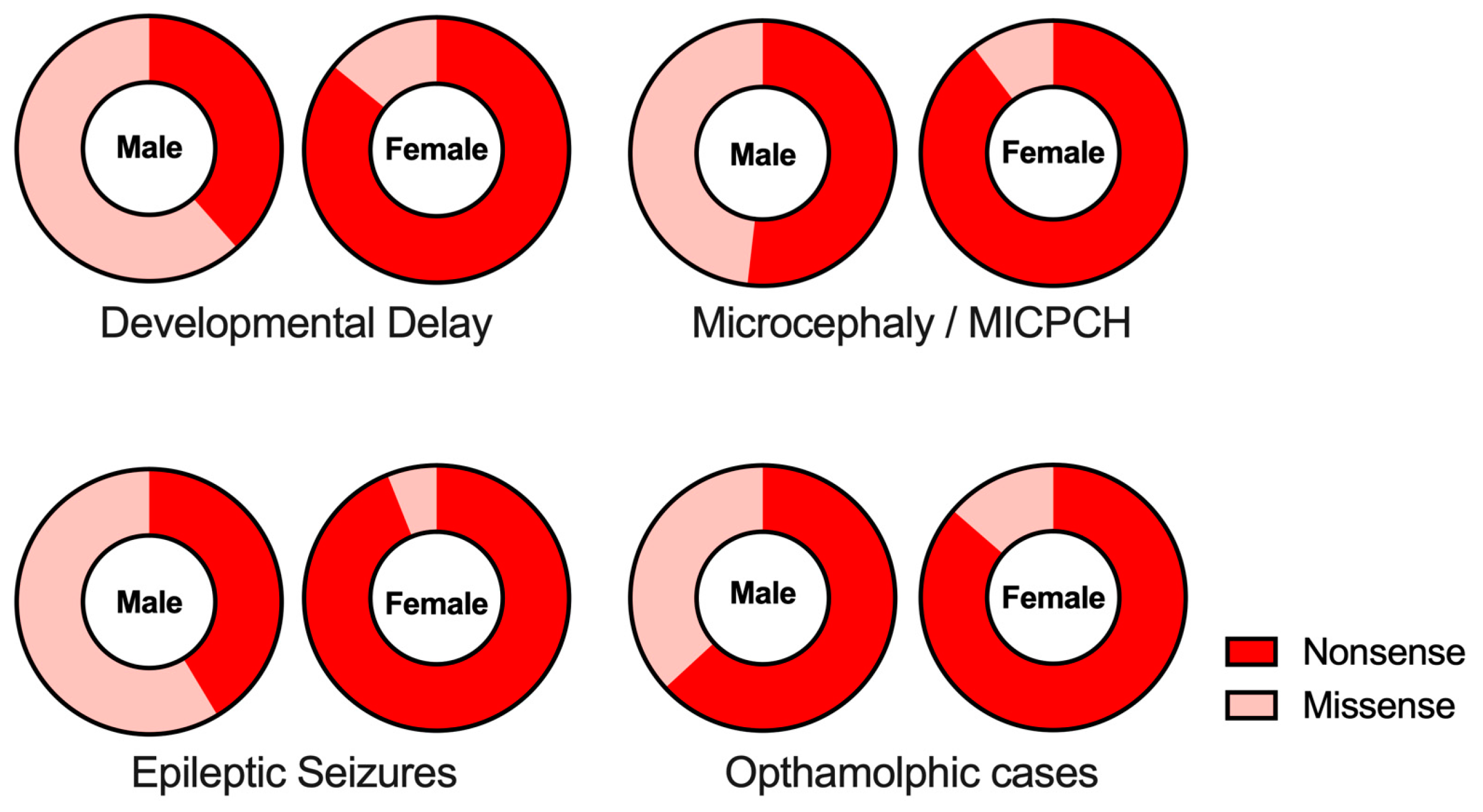
4. Phenotypes and Functional Domains of CASK
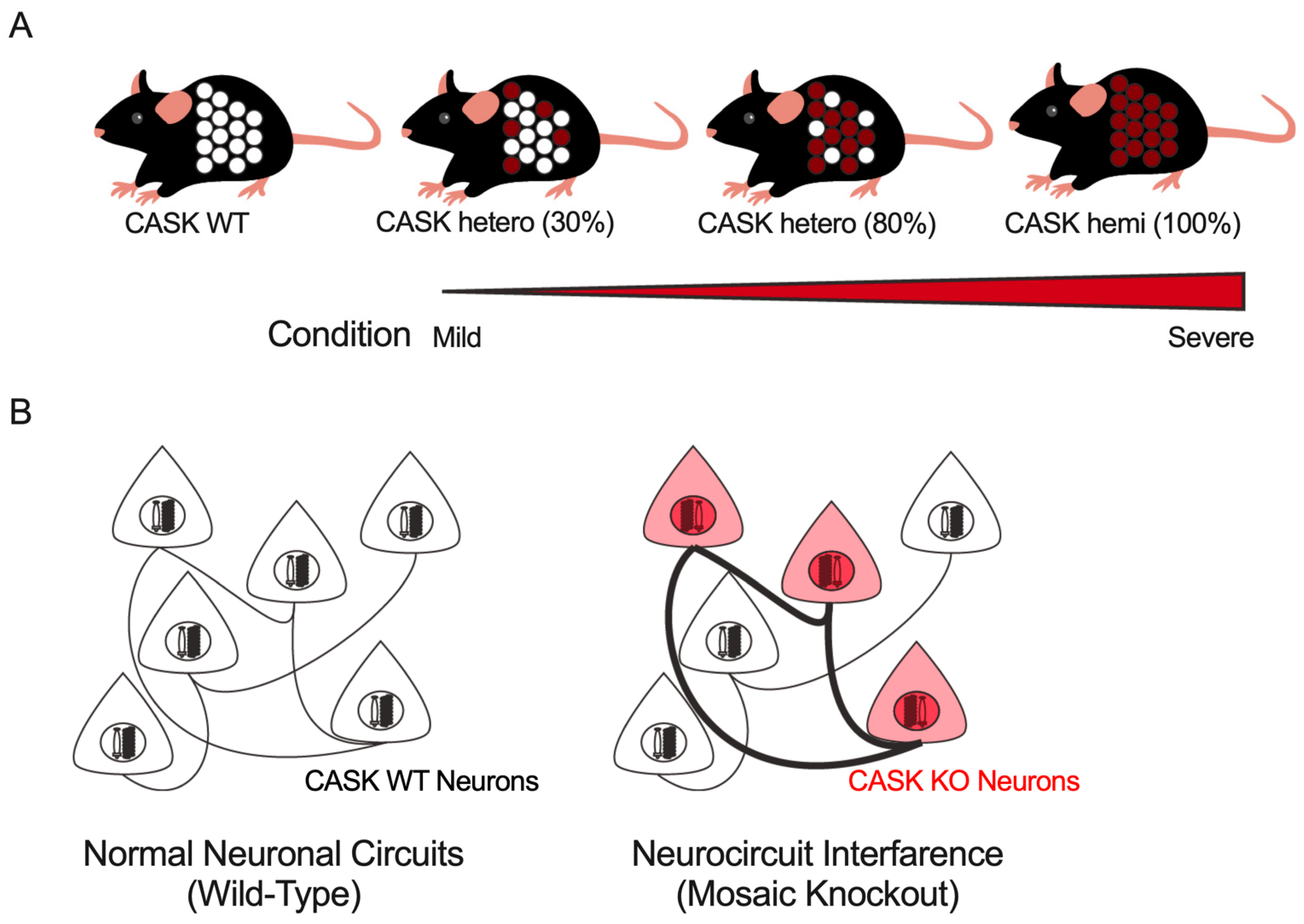
5. Unresolved Questions on the Pathophysiology of CASK-Related Disorders
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohen, A.R.; Woods, D.F.; Marfatia, S.M.; Walther, Z.; Chishti, A.H.; Anderson, J.M. Human CASK/LIN-2 binds syndecan-2 and protein 4.1 and localizes to the basolateral membrane of epithelial cells. J. Cell Biol. 1998, 142, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Dimitratos, S.D.; Stathakis, D.G.; Nelson, C.A.; Woods, D.F.; Bryant, P.J. The location of human CASK at Xp11.4 identifies this gene as a candidate for X-linked optic atrophy. Genomics 1998, 51, 308–309. [Google Scholar] [CrossRef] [PubMed]
- Froyen, G.; Van Esch, H.; Bauters, M.; Hollanders, K.; Frints, S.G.; Vermeesch, J.R.; Devriendt, K.; Fryns, J.P.; Marynen, P. Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: Important role for increased gene dosage of XLMR genes. Hum. Mutat. 2007, 28, 1034–1042. [Google Scholar] [CrossRef]
- Najm, J.; Horn, D.; Wimplinger, I.; Golden, J.A.; Chizhikov, V.V.; Sudi, J.; Christian, S.L.; Ullmann, R.; Kuechler, A.; Haas, C.A.; et al. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet. 2008, 40, 1065–1067. [Google Scholar] [CrossRef] [PubMed]
- Burglen, L.; Chantot-Bastaraud, S.; Garel, C.; Milh, M.; Touraine, R.; Zanni, G.; Petit, F.; Afenjar, A.; Goizet, C.; Barresi, S.; et al. Spectrum of pontocerebellar hypoplasia in 13 girls and boys with CASK mutations: Confirmation of a recognizable phenotype and first description of a male mosaic patient. Orphanet. J. Rare Dis. 2012, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Kato, M.; Osaka, H.; Moriyama, N.; Horita, H.; Nishiyama, K.; Yoneda, Y.; Kondo, Y.; Tsurusaki, Y.; Doi, H.; et al. CASK aberrations in male patients with Ohtahara syndrome and cerebellar hypoplasia. Epilepsia 2012, 53, 1441–1449. [Google Scholar] [CrossRef]
- Takanashi, J.; Okamoto, N.; Yamamoto, Y.; Hayashi, S.; Arai, H.; Takahashi, Y.; Maruyama, K.; Mizuno, S.; Shimakawa, S.; Ono, H.; et al. Clinical and radiological features of Japanese patients with a severe phenotype due to CASK mutations. Am. J. Med. Genet. A 2012, 158A, 3112–3118. [Google Scholar] [CrossRef]
- Nakamura, K.; Nishiyama, K.; Kodera, H.; Nakashima, M.; Tsurusaki, Y.; Miyake, N.; Matsumoto, N.; Saitsu, H.; Jinnou, H.; Ohki, S.; et al. A de novo CASK mutation in pontocerebellar hypoplasia type 3 with early myoclonic epilepsy and tetralogy of Fallot. Brain Dev. 2014, 36, 272–273. [Google Scholar] [CrossRef]
- Nakajiri, T.; Kobayashi, K.; Okamoto, N.; Oka, M.; Miya, F.; Kosaki, K.; Yoshinaga, H. Late-onset epileptic spasms in a female patient with a CASK mutation. Brain Dev. 2015, 37, 919–923. [Google Scholar] [CrossRef]
- Giacomini, T.; Nuovo, S.; Zanni, G.; Mancardi, M.M.; Cusmai, R.; Pepi, C.; Bertini, E.; Valente, E.M.; Battini, R.; Ferrari, A.; et al. CASK related disorder: Epilepsy and developmental outcome. Eur. J. Paediatr. Neurol. 2021, 31, 61–69. [Google Scholar] [CrossRef]
- Moog, U.; Kutsche, K.; Kortüm, F.; Chilian, B.; Bierhals, T.; Apeshiotis, N.; Balg, S.; Chassaing, N.; Coubes, C.; Das, S.; et al. Phenotypic spectrum associated with CASK loss-of-function mutations. J. Med. Genet. 2011, 48, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Piluso, G.; D’Amico, F.; Saccone, V.; Bismuto, E.; Rotundo, I.L.; Di Domenico, M.; Aurino, S.; Schwartz, C.E.; Neri, G.; Nigro, V. A missense mutation in CASK causes FG syndrome in an Italian family. Am. J. Hum. Genet. 2009, 84, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Dunn, P.; Prigatano, G.P.; Szelinger, S.; Roth, J.; Siniard, A.L.; Claasen, A.M.; Richholt, R.F.; De Both, M.; Corneveaux, J.J.; Moskowitz, A.M.; et al. A de novo splice site mutation in CASK causes FG syndrome-4 and congenital nystagmus. Am. J. Med. Genet. A 2017, 173, 611–617. [Google Scholar] [CrossRef]
- Hackett, A.; Tarpey, P.S.; Licata, A.; Cox, J.; Whibley, A.; Boyle, J.; Rogers, C.; Grigg, J.; Partington, M.; Stevenson, R.E.; et al. CASK mutations are frequent in males and cause X-linked nystagmus and variable XLMR phenotypes. Eur. J. Hum. Genet. 2010, 18, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.; Mastropasqua, F.; Reising, J.P.; Maier, S.; Ho, M.L.; Rabkina, I.; Li, D.; Neufeld, J.; Ballenberger, L.; Myers, L.; et al. Presynaptic dysfunction in CASK-related neurodevelopmental disorders. Transl. Psychiatry 2020, 10, 312. [Google Scholar] [CrossRef]
- Mustroph, J.; Sag, C.M.; Bähr, F.; Schmidtmann, A.L.; Gupta, S.N.; Dietz, A.; Islam, M.M.T.; Lücht, C.; Beuthner, B.E.; Pabel, S.; et al. Loss of CASK Accelerates Heart Failure Development. Circ. Res. 2021, 128, 1139–1155. [Google Scholar] [CrossRef]
- Wei, J.L.; Fu, Z.X.; Fang, M.; Zhou, Q.Y.; Zhao, Q.N.; Guo, J.B.; Lu, W.D.; Wang, H. High expression of CASK correlates with progression and poor prognosis of colorectal cancer. Tumor Biol. 2014, 35, 9185–9194. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, G.; Yin, C.; Jin, W.; Zhang, G. Down-regulation of miR-203 induced by Helicobacter pylori infection promotes the proliferation and invasion of gastric cancer by targeting CASK. Oncotarget 2014, 5, 11631–11640. [Google Scholar] [CrossRef]
- Ding, B.; Bao, C.; Jin, L.; Xu, L.; Fan, W.; Lou, W. CASK Silence Overcomes Sorafenib Resistance of Hepatocellular Carcinoma through Activating Apoptosis and Autophagic Cell Death. Front. Oncol. 2021, 11, 681683. [Google Scholar] [CrossRef]
- Qu, J.; Zhou, Y.; Li, Y.; Yu, J.; Wang, W. CASK regulates Notch pathway and functions as a tumor promoter in pancreatic cancer. Arch. Biochem. Biophys. 2021, 701, 108789. [Google Scholar] [CrossRef]
- Hata, Y.; Butz, S.; Südhof, T.C. CASK: A novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J. Neurosci. 1996, 16, 2488–2494. [Google Scholar] [CrossRef] [PubMed]
- Baines, A.J. Caenorhabditis elegans LIN-2A and mammalian neuronal CASK are prototypical members of a subfamily of MAGUKs (membrane-associated guanylate kinases) characterized by a common kinase-like domain and a guanylate kinase domain predicted to bind ATP. Biochem. J. 1996, 320 Pt 2, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Dimitratos, S.D.; Woods, D.F.; Bryant, P.J. Camguk, Lin-2, and CASK: Novel membrane-associated guanylate kinase homologs that also contain CaM kinase domains. Mech. Dev. 1997, 63, 127–130. [Google Scholar] [CrossRef]
- Mukherjee, K.; Sharma, M.; Urlaub, H.; Bourenkov, G.P.; Jahn, R.; Südhof, T.C.; Wahl, M.C. CASK Functions as a Mg2+-independent neurexin kinase. Cell 2008, 133, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Zheng, S.; Spangler, S.A.; Yu, C.; Hoogenraad, C.C.; Zhang, M. Liprin-mediated large signaling complex organization revealed by the liprin-α/CASK and liprin-α/liprin-β complex structures. Mol. Cell 2011, 43, 586–598. [Google Scholar] [CrossRef]
- Spangler, S.A.; Schmitz, S.K.; Kevenaar, J.T.; de Graaff, E.; de Wit, H.; Demmers, J.; Toonen, R.F.; Hoogenraad, C.C. Liprin-α2 promotes the presynaptic recruitment and turnover of RIM1/CASK to facilitate synaptic transmission. J. Cell Biol. 2013, 201, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Leonoudakis, D.; Conti, L.R.; Radeke, C.M.; McGuire, L.M.; Vandenberg, C.A. A multiprotein trafficking complex composed of SAP97, CASK, Veli, and Mint1 is associated with inward rectifier Kir2 potassium channels. J. Biol. Chem. 2004, 279, 19051–19063. [Google Scholar] [CrossRef]
- Hong, C.J.; Hsueh, Y.P. CASK associates with glutamate receptor interacting protein and signaling molecules. Biochem. Biophys. Res. Commun. 2006, 351, 771–776. [Google Scholar] [CrossRef]
- Stafford, R.L.; Ear, J.; Knight, M.J.; Bowie, J.U. The molecular basis of the Caskin1 and Mint1 interaction with CASK. J. Mol. Biol. 2011, 412, 3–13. [Google Scholar] [CrossRef]
- Lee, S.; Fan, S.; Makarova, O.; Straight, S.; Margolis, B. A novel and conserved protein-protein interaction domain of mammalian Lin-2/CASK binds and recruits SAP97 to the lateral surface of epithelia. Mol. Cell. Biol. 2002, 22, 1778–1791. [Google Scholar] [CrossRef]
- Jeyifous, O.; Waites, C.L.; Specht, C.G.; Fujisawa, S.; Schubert, M.; Lin, E.I.; Marshall, J.; Aoki, C.; de Silva, T.; Montgomery, J.M.; et al. SAP97 and CASK mediate sorting of NMDA receptors through a previously unknown secretory pathway. Nat. Neurosci. 2009, 12, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Olsen, O.; Liu, H.; Wade, J.B.; Merot, J.; Welling, P.A. Basolateral membrane expression of the Kir 2.3 channel is coordinated by PDZ interaction with Lin-7/CASK complex. Am. J. Physiol. Cell Physiol. 2002, 282, C183–C195. [Google Scholar] [CrossRef] [PubMed]
- Alewine, C.; Kim, B.Y.; Hegde, V.; Welling, P.A. Lin-7 targets the Kir 2.3 channel on the basolateral membrane via a L27 domain interaction with CASK. Am. J. Physiol. Cell Physiol. 2007, 293, C1733–C1741. [Google Scholar] [CrossRef]
- Hsueh, Y.P.; Yang, F.C.; Kharazia, V.; Naisbitt, S.; Cohen, A.R.; Weinberg, R.J.; Sheng, M. Direct interaction of CASK/LIN-2 and syndecan heparan sulfate proteoglycan and their overlapping distribution in neuronal synapses. J. Cell Biol. 1998, 142, 139–151. [Google Scholar] [CrossRef]
- Hsueh, Y.P.; Sheng, M. Regulated expression and subcellular localization of syndecan heparan sulfate proteoglycans and the syndecan-binding protein CASK/LIN-2 during rat brain development. J. Neurosci. 1999, 19, 7415–7425. [Google Scholar] [CrossRef] [PubMed]
- Gomez, A.M.; Traunmüller, L.; Scheiffele, P. Neurexins: Molecular codes for shaping neuronal synapses. Nat. Rev. Neurosci. 2021, 22, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Maximov, A.; Südhof, T.C.; Bezprozvanny, I. Association of neuronal calcium channels with modular adaptor proteins. J. Biol. Chem. 1999, 274, 24453–24456. [Google Scholar] [CrossRef]
- Maximov, A.; Bezprozvanny, I. Synaptic targeting of N-type calcium channels in hippocampal neurons. J. Neurosci. 2002, 22, 6939–6952. [Google Scholar] [CrossRef]
- Hsueh, Y.P.; Wang, T.F.; Yang, F.C.; Sheng, M. Nuclear translocation and transcription regulation by the membrane-associated guanylate kinase CASK/LIN-2. Nature 2000, 404, 298–302. [Google Scholar] [CrossRef]
- Wang, G.S.; Hong, C.J.; Yen, T.Y.; Huang, H.Y.; Ou, Y.; Huang, T.N.; Jung, W.G.; Kuo, T.Y.; Sheng, M.; Wang, T.F.; et al. Transcriptional modification by a CASK-interacting nucleosome assembly protein. Neuron 2004, 42, 113–128. [Google Scholar] [CrossRef]
- Lin, C.W.; Huang, T.N.; Wang, G.S.; Kuo, T.Y.; Yen, T.Y.; Hsueh, Y.P. Neural activity- and development-dependent expression and distribution of CASK interacting nucleosome assembly protein in mouse brain. J. Comp. Neurol. 2006, 494, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lu, J.; Yang, C.; Wang, X.; Cheng, L.; Hu, G.; Sun, Y.; Zhang, X.; Wu, M.; Liu, Z. CASK and its target gene Reelin were co-upregulated in human esophageal carcinoma. Cancer Lett. 2002, 179, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.N.; Hsueh, Y.P. CASK point mutation regulates protein-protein interactions and NR2b promoter activity. Biochem. Biophys. Res. Commun. 2009, 382, 219–222. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Pan, Y.E.; Tibbe, D.; Harms, F.L.; Reißner, C.; Becker, K.; Dingmann, B.; Mirzaa, G.; Kattentidt-Mouravieva, A.A.; Shoukier, M.; Aggarwal, S.; et al. Missense mutations in CASK, coding for the calcium-/calmodulin-dependent serine protein kinase, interfere with neurexin binding and neurexin-induced oligomerization. J. Neurochem. 2021, 157, 1331–1350. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef]
- Hayashi, S.; Mizuno, S.; Migita, O.; Okuyama, T.; Makita, Y.; Hata, A.; Imoto, I.; Inazawa, J. The CASK gene harbored in a deletion detected by array-CGH as a potential candidate for a gene causative of X-linked dominant mental retardation. Am. J. Med. Genet. A 2008, 146A, 2145–2151. [Google Scholar] [CrossRef]
- Tarpey, P.S.; Smith, R.; Pleasance, E.; Whibley, A.; Edkins, S.; Hardy, C.; O’Meara, S.; Latimer, C.; Dicks, E.; Menzies, A.; et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009, 41, 535–543. [Google Scholar] [CrossRef]
- Hayashi, S.; Okamoto, N.; Chinen, Y.; Takanashi, J.; Makita, Y.; Hata, A.; Imoto, I.; Inazawa, J. Novel intragenic duplications and mutations of CASK in patients with mental retardation and microcephaly with pontine and cerebellar hypoplasia (MICPCH). Hum. Genet. 2012, 131, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Valayannopoulos, V.; Michot, C.; Rodriguez, D.; Hubert, L.; Saillour, Y.; Labrune, P.; de Laveaucoupet, J.; Brunelle, F.; Amiel, J.; Lyonnet, S.; et al. Mutations of TSEN and CASK genes are prevalent in pontocerebellar hypoplasias type 2 and 4. Brain 2012, 135, e199; author reply e200. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.L.; Lachance, M.; Hamdan, F.F.; Carmant, L.; Lortie, A.; Diadori, P.; Major, P.; Meijer, I.A.; Lemyre, E.; Cossette, P.; et al. The genetic landscape of infantile spasms. Hum. Mol. Genet. 2014, 23, 4846–4858. [Google Scholar] [CrossRef] [PubMed]
- Moog, U.; Bierhals, T.; Brand, K.; Bautsch, J.; Biskup, S.; Brune, T.; Denecke, J.; de Die-Smulders, C.E.; Evers, C.; Hempel, M.; et al. Phenotypic and molecular insights into CASK-related disorders in males. Orphanet. J. Rare Dis. 2015, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Rump, P.; Jazayeri, O.; van Dijk-Bos, K.K.; Johansson, L.F.; van Essen, A.J.; Verheij, J.B.; Veenstra-Knol, H.E.; Redeker, E.J.; Mannens, M.M.; Swertz, M.A.; et al. Whole-exome sequencing is a powerful approach for establishing the etiological diagnosis in patients with intellectual disability and microcephaly. BMC Med. Genom. 2016, 9, 7. [Google Scholar] [CrossRef]
- Rivas, L.; Blanco, Ó.; Torreira, C.; Repáraz, A.; Melcón, C.; Amado, A. Pontocerebellar hypoplasia secondary to CASK gene deletion: Case report. Rev. Chil. Pediatr. 2017, 88, 529–533. [Google Scholar] [CrossRef]
- Hayashi, S.; Uehara, D.T.; Tanimoto, K.; Mizuno, S.; Chinen, Y.; Fukumura, S.; Takanashi, J.I.; Osaka, H.; Okamoto, N.; Inazawa, J. Comprehensive investigation of CASK mutations and other genetic etiologies in 41 patients with intellectual disability and microcephaly with pontine and cerebellar hypoplasia (MICPCH). PLoS ONE 2017, 12, e0181791. [Google Scholar] [CrossRef]
- Popp, B.; Ekici, A.B.; Thiel, C.T.; Hoyer, J.; Wiesener, A.; Kraus, C.; Reis, A.; Zweier, C. Exome Pool-Seq in neurodevelopmental disorders. Eur. J. Hum. Genet. 2017, 25, 1364–1376. [Google Scholar] [CrossRef]
- DeLuca, S.C.; Wallace, D.A.; Trucks, M.R.; Mukherjee, K. A clinical series using intensive neurorehabilitation to promote functional motor and cognitive skills in three girls with CASK mutation. BMC Res. Notes 2017, 10, 743. [Google Scholar] [CrossRef]
- Seto, T.; Hamazaki, T.; Nishigaki, S.; Kudo, S.; Shintaku, H.; Ondo, Y.; Shimojima, K.; Yamamoto, T. A novel CASK mutation identified in siblings exhibiting developmental disorders with/without microcephaly. Intractable Rare Dis. Res. 2017, 6, 177–182. [Google Scholar] [CrossRef]
- Muthusamy, B.; Selvan, L.D.N.; Nguyen, T.T.; Manoj, J.; Stawiski, E.W.; Jaiswal, B.S.; Wang, W.; Raja, R.; Ramprasad, V.L.; Gupta, R.; et al. Next-Generation Sequencing Reveals Novel Mutations in X-linked Intellectual Disability. OMICS 2017, 21, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Bozarth, X.; Foss, K.; Mefford, H.C. A de novo in-frame deletion of CASK gene causes early onset infantile spasms and supratentorial cerebral malformation in a female patient. Am. J. Med. Genet. A 2018, 176, 2425–2429. [Google Scholar] [CrossRef]
- LaConte, L.E.W.; Chavan, V.; Elias, A.F.; Hudson, C.; Schwanke, C.; Styren, K.; Shoof, J.; Kok, F.; Srivastava, S.; Mukherjee, K. Two microcephaly-associated novel missense mutations in CASK specifically disrupt the CASK-neurexin interaction. Hum. Genet. 2018, 137, 231–246. [Google Scholar] [CrossRef] [PubMed]
- LaConte, L.E.W.; Chavan, V.; DeLuca, S.; Rubin, K.; Malc, J.; Berry, S.; Gail Summers, C.; Mukherjee, K. An N-terminal heterozygous missense CASK mutation is associated with microcephaly and bilateral retinal dystrophy plus optic nerve atrophy. Am. J. Med. Genet. A 2019, 179, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Cristofoli, F.; Devriendt, K.; Davis, E.E.; Van Esch, H.; Vermeesch, J.R. Novel CASK mutations in cases with syndromic microcephaly. Hum. Mutat. 2018, 39, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Hauer, N.N.; Popp, B.; Schoeller, E.; Schuhmann, S.; Heath, K.E.; Hisado-Oliva, A.; Klinger, P.; Kraus, C.; Trautmann, U.; Zenker, M.; et al. Clinical relevance of systematic phenotyping and exome sequencing in patients with short stature. Genet. Med. 2018, 20, 630–638. [Google Scholar] [CrossRef]
- Murakami, H.; Kimura, Y.; Enomoto, Y.; Tsurusaki, Y.; Akahira-Azuma, M.; Kuroda, Y.; Tsuji, M.; Goto, T.; Kurosawa, K. Discordant phenotype caused by CASK mutation in siblings with NF1. Hum. Genome Var. 2019, 6, 20. [Google Scholar] [CrossRef]
- Zhang, K.; Yuan, Q.; Xie, J.; Yuan, L.; Wang, Y. PPAR-γ activation increases insulin secretion independent of CASK in INS-1 cells. Acta Biochim. Biophys. Sin. 2019, 51, 715–722. [Google Scholar] [CrossRef]
- Aspromonte, M.C.; Bellini, M.; Gasparini, A.; Carraro, M.; Bettella, E.; Polli, R.; Cesca, F.; Bigoni, S.; Boni, S.; Carlet, O.; et al. Characterization of intellectual disability and autism comorbidity through gene panel sequencing. Hum. Mutat. 2019, 40, 1346–1363. [Google Scholar] [CrossRef]
- Rochtus, A.; Olson, H.E.; Smith, L.; Keith, L.G.; El Achkar, C.; Taylor, A.; Mahida, S.; Park, M.; Kelly, M.; Shain, C.; et al. Genetic diagnoses in epilepsy: The impact of dynamic exome analysis in a pediatric cohort. Epilepsia 2020, 61, 249–258. [Google Scholar] [CrossRef]
- Ibarluzea, N.; Hoz, A.B.; Villate, O.; Llano, I.; Ocio, I.; Martí, I.; Guitart, M.; Gabau, E.; Andrade, F.; Gener, B.; et al. Targeted Next-Generation Sequencing in Patients with Suggestive X-Linked Intellectual Disability. Genes 2020, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.X.; Ma, H.X.; Zhang, Y.X.; Chen, Z.H.; Zhai, Q.X. Whole-Exome Sequencing for Identifying Genetic Causes of Intellectual Developmental Disorders. Int. J. Gen. Med. 2021, 14, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Dubbs, H.; Ortiz-Gonzalez, X.; Marsh, E.D. Pathogenic variants in CASK: Expanding the genotype-phenotype correlations. Am. J. Med. Genet. A 2022, 188, 2617–2626. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Zhang, Y.; Yang, W.; Yang, L.; Wang, R.; Xu, M.; Sun, L.; Zhang, B.; Cui, X. Case report: A novel CASK mutation in a Chinese female child with microcephaly with pontine and cerebellar hypoplasia. Front. Genet. 2022, 13, 856636. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Jia, P.; Yao, Y.; Zhu, F. Case Report: Identification of a novel CASK missense variant in a Chinese family with MICPCH. Front. Genet. 2022, 13, 933785. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Jiang, C.; Li, J.; Zhang, G.; Shen, Y.; Wang, J. A novel missense variant in the CASK gene causes intellectual developmental disorder and microcephaly with pontine and cerebellar hypoplasia. BMC Med. Genom. 2022, 15, 127. [Google Scholar] [CrossRef]
- Zhang, Y.; Nie, Y.; Mu, Y.; Zheng, J.; Xu, X.; Zhang, F.; Shu, J.; Liu, Y. A de novo variant in CASK gene causing intellectual disability and brain hypoplasia: A case report and literature review. Ital. J. Pediatr. 2022, 48, 73. [Google Scholar] [CrossRef]
- Yang, K.; Lin, L.; Yuan, F.; Li, X.; Liu, Z.; Lan, X.; Wang, Y.; Ren, Y.; Li, J.; Chen, Y. Two heterozygous mutations in the calcium/calmodulin-dependent serine protein kinase gene (CASK) in cases with developmental disorders. Mol. Genet. Genom. Med. 2022, 10, e2065. [Google Scholar] [CrossRef]
- Patel, P.A.; Hegert, J.V.; Cristian, I.; Kerr, A.; LaConte, L.E.W.; Fox, M.A.; Srivastava, S.; Mukherjee, K. Complete loss of the X-linked gene CASK causes severe cerebellar degeneration. J. Med. Genet. 2022, 59, 1044–1057. [Google Scholar] [CrossRef]
- Lai, D.; Gade, M.; Yang, E.; Koh, H.Y.; Lu, J.; Walley, N.M.; Buckley, A.F.; Sands, T.T.; Akman, C.I.; Mikati, M.A.; et al. Somatic variants in diverse genes leads to a spectrum of focal cortical malformations. Brain 2022, 145, 2704–2720. [Google Scholar] [CrossRef]
- Tibbe, D.; Ferle, P.; Krisp, C.; Nampoothiri, S.; Mirzaa, G.; Assaf, M.; Parikh, S.; Kutsche, K.; Kreienkamp, H.J. Regulation of Liprin-α phase separation by CASK is disrupted by a mutation in its CaM kinase domain. Life Sci. Alliance 2022, 5, e202201512. [Google Scholar] [CrossRef] [PubMed]
- Abe-Hatano, C.; Yokoi, T.; Ida, K.; Kurosawa, K. Mosaicism of a Truncating Variant of CASK Causes Congenital Heart Disease and Neurodevelopmental Disorder. Mol. Syndromol. 2023, 13, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Long, S.; Zhou, H.; Li, S.; Wang, T.; Ma, Y.; Li, C.; Zhou, Y.; Zhou, S.; Wu, B.; Wang, Y. The Clinical and Genetic Features of Co-occurring Epilepsy and Autism Spectrum Disorder in Chinese Children. Front. Neurol. 2019, 10, 505. [Google Scholar] [CrossRef] [PubMed]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017, 542, 433–438. [CrossRef]
- Tabuchi, K.; Blundell, J.; Etherton, M.R.; Hammer, R.E.; Liu, X.; Powell, C.M.; Südhof, T.C. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 2007, 318, 71–76. [Google Scholar] [CrossRef]
- Jamain, S.; Quach, H.; Betancur, C.; Råstam, M.; Colineaux, C.; Gillberg, I.C.; Soderstrom, H.; Giros, B.; Leboyer, M.; Gillberg, C.; et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003, 34, 27–29. [Google Scholar] [CrossRef]
- Hansen, J.; Snow, C.; Tuttle, E.; Ghoneim, D.H.; Yang, C.S.; Spencer, A.; Gunter, S.A.; Smyser, C.D.; Gurnett, C.A.; Shinawi, M.; et al. De novo mutations in SIK1 cause a spectrum of developmental epilepsies. Am. J. Hum. Genet. 2015, 96, 682–690. [Google Scholar] [CrossRef]
- Badawi, M.; Mori, T.; Kurihara, T.; Yoshizawa, T.; Nohara, K.; Kouyama-Suzuki, E.; Yanagawa, T.; Shirai, Y.; Tabuchi, K. Risperidone Mitigates Enhanced Excitatory Neuronal Function and Repetitive Behavior Caused by an ASD-Associated Mutation of SIK1. Front. Mol. Neurosci. 2021, 14, 706494. [Google Scholar] [CrossRef]
- Borsani, G.; Tonlorenzi, R.; Simmler, M.C.; Dandolo, L.; Arnaud, D.; Capra, V.; Grompe, M.; Pizzuti, A.; Muzny, D.; Lawrence, C.; et al. Characterization of a murine gene expressed from the inactive X chromosome. Nature 1991, 351, 325–329. [Google Scholar] [CrossRef]
- Huynh, K.D.; Lee, J.T. X-chromosome inactivation: A hypothesis linking ontogeny and phylogeny. Nat. Rev. Genet. 2005, 6, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Kasem, E.A.; Suzuki-Kouyama, E.; Cao, X.; Li, X.; Kurihara, T.; Uemura, T.; Yanagawa, T.; Tabuchi, K. Deficiency of calcium/calmodulin-dependent serine protein kinase disrupts the excitatory-inhibitory balance of synapses by down-regulating GluN2B. Mol. Psychiatry 2019, 24, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; McMillan, R.; Willis, J.; Clark, H.; Chavan, V.; Liang, C.; Zhang, H.; Hulver, M.; Mukherjee, K. X-linked intellectual disability gene CASK regulates postnatal brain growth in a non-cell autonomous manner. Acta Neuropathol. Commun. 2016, 4, 30. [Google Scholar] [CrossRef]
- Guo, Q.; Kouyama-Suzuki, E.; Shirai, Y.; Cao, X.; Yanagawa, T.; Mori, T.; Tabuchi, K. Structural Analysis Implicates CASK-Liprin-α2 Interaction in Cerebellar Granular Cell Death in MICPCH Syndrome. Cells 2023, 12, 1177. [Google Scholar] [CrossRef]
- Uemura, T.; Suzuki-Kouyama, E.; Kawase, S.; Kurihara, T.; Yasumura, M.; Yoshida, T.; Fukai, S.; Yamazaki, M.; Fei, P.; Abe, M.; et al. Neurexins play a crucial role in cerebellar granule cell survival by organizing autocrine machinery for neurotrophins. Cell Rep. 2022, 39, 110624. [Google Scholar] [CrossRef]
- Mori, T.; Shimizu, K.; Hayashi, M. Differential expression patterns of TrkB ligands in the macaque monkey brain. Neuroreport 2004, 15, 2507–2511. [Google Scholar] [CrossRef]
- Schwartz, P.M.; Borghesani, P.R.; Levy, R.L.; Pomeroy, S.L.; Segal, R.A. Abnormal cerebellar development and foliation in BDNF−/− mice reveals a role for neurotrophins in CNS patterning. Neuron 1997, 19, 269–281. [Google Scholar] [CrossRef]
- Borghesani, P.R.; Peyrin, J.M.; Klein, R.; Rubin, J.; Carter, A.R.; Schwartz, P.M.; Luster, A.; Corfas, G.; Segal, R.A. BDNF stimulates migration of cerebellar granule cells. Development 2002, 129, 1435–1442. [Google Scholar] [CrossRef]
- LaConte, L.E.; Chavan, V.; Liang, C.; Willis, J.; Schönhense, E.M.; Schoch, S.; Mukherjee, K. CASK stabilizes neurexin and links it to liprin-α in a neuronal activity-dependent manner. Cell. Mol. Life Sci. 2016, 73, 3599–3621. [Google Scholar] [CrossRef]
- Lemke, J.R.; Hendrickx, R.; Geider, K.; Laube, B.; Schwake, M.; Harvey, R.J.; James, V.M.; Pepler, A.; Steiner, I.; Hörtnagel, K.; et al. GRIN2B mutations in West syndrome and intellectual disability with focal epilepsy. Ann. Neurol. 2014, 75, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Platzer, K.; Yuan, H.; Schütz, H.; Winschel, A.; Chen, W.; Hu, C.; Kusumoto, H.; Heyne, H.O.; Helbig, K.L.; Tang, S.; et al. encephalopathy: Novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J. Med. Genet. 2017, 54, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.Y.; Yang, X.F.; Tomonoh, Y.; Hu, L.Y.; Ju, J.; Hirose, S.; Zou, L.P. Development of a mouse model of infantile spasms induced by N-methyl-D-aspartate. Epilepsy Res. 2015, 118, 29–33. [Google Scholar] [CrossRef]
- Pang, B.; Mori, T.; Badawi, M.; Zhou, M.; Guo, Q.; Suzuki-Kouyama, E.; Yanagawa, T.; Shirai, Y.; Tabuchi, K. An Epilepsy-Associated Mutation of Salt-Inducible Kinase 1 Increases the Susceptibility to Epileptic Seizures and Interferes with Adrenocorticotropic Hormone Therapy for Infantile Spasms in Mice. Int. J. Mol. Sci. 2022, 23, 7927. [Google Scholar] [CrossRef] [PubMed]
- Velísek, L.; Jehle, K.; Asche, S.; Velísková, J. Model of infantile spasms induced by N-methyl-D-aspartic acid in prenatally impaired brain. Ann. Neurol. 2007, 61, 109–119. [Google Scholar] [CrossRef]
- Atasoy, D.; Schoch, S.; Ho, A.; Nadasy, K.A.; Liu, X.; Zhang, W.; Mukherjee, K.; Nosyreva, E.D.; Fernandez-Chacon, R.; Missler, M.; et al. Deletion of CASK in mice is lethal and impairs synaptic function. Proc. Natl. Acad. Sci. USA 2007, 104, 2525–2530. [Google Scholar] [CrossRef] [PubMed]
- Gafner, M.; Boltshauser, E.; D’Abrusco, F.; Battini, R.; Romaniello, R.; D’Arrigo, S.; Zanni, G.; Leibovitz, Z.; Yosovich, K.; Lerman-Sagie, T.; et al. Expanding the natural history of CASK-related disorders to the prenatal period. Dev. Med. Child. Neurol. 2023, 65, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Zoghbi, H.Y.; Bear, M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, a009886. [Google Scholar] [CrossRef]
- Depienne, C.; Bouteiller, D.; Keren, B.; Cheuret, E.; Poirier, K.; Trouillard, O.; Benyahia, B.; Quelin, C.; Carpentier, W.; Julia, S.; et al. Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet. 2009, 5, e1000381. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutations | Pathogenic | Likely Pathogenic | Benign 1 | Uncertain Significance 2 |
|---|---|---|---|---|
| Frameshift | 30 | 6 | 0 | 1 |
| Missense * | 11 | 20 | 14 | 180 |
| Nonsense | 40 | 3 | 0 | 0 |
| Phenotypes | Gender | Severe | Mild/No | NA 1 |
|---|---|---|---|---|
| Intellectual Disability | Male | 58 | 4 | 3 |
| Female | 124 | 5 | 3 | |
| Microcephaly/MICPCH | Male | 35 | 11 | 19 |
| Female | 107 | 15 | 10 | |
| Epilepsy * | Male | 33 | 28 | 4 |
| Female | 44 | 78 | 10 |
| Phenotypes | Mutations | Male | Female | Total |
|---|---|---|---|---|
| Intellectual disability ** | Nonsense 1 | 24 | 109 | 133 |
| Missense | 34 | 15 | 49 | |
| Microcephaly/MICPCH ** | Nonsense 1 | 19 | 98 | 117 |
| Missense | 14 | 9 | 23 | |
| Epileptic seizures ** | Nonsense 1 | 16 | 40 | 56 |
| Missense | 17 | 2 | 19 | |
| Ophthalmological anomalies * | Nonsense 1 | 16 | 38 | 64 |
| Missense | 11 | 7 | 18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mori, T.; Zhou, M.; Tabuchi, K. Diverse Clinical Phenotypes of CASK-Related Disorders and Multiple Functional Domains of CASK Protein. Genes 2023, 14, 1656. https://doi.org/10.3390/genes14081656
Mori T, Zhou M, Tabuchi K. Diverse Clinical Phenotypes of CASK-Related Disorders and Multiple Functional Domains of CASK Protein. Genes. 2023; 14(8):1656. https://doi.org/10.3390/genes14081656
Chicago/Turabian StyleMori, Takuma, Mengyun Zhou, and Katsuhiko Tabuchi. 2023. "Diverse Clinical Phenotypes of CASK-Related Disorders and Multiple Functional Domains of CASK Protein" Genes 14, no. 8: 1656. https://doi.org/10.3390/genes14081656
APA StyleMori, T., Zhou, M., & Tabuchi, K. (2023). Diverse Clinical Phenotypes of CASK-Related Disorders and Multiple Functional Domains of CASK Protein. Genes, 14(8), 1656. https://doi.org/10.3390/genes14081656