Abstract
Snijders Blok–Campeau syndrome (SNIBCPS, OMIM# 618205) is an extremely infrequent disease with only approximately 60 cases reported so far. SNIBCPS belongs to the group of neurodevelopmental disorders (NDDs). Clinical features of patients with SNIBCPS include global developmental delay, intellectual disability, speech and language difficulties and behavioral disorders like autism spectrum disorder. In addition, patients with SNIBCPS exhibit typical dysmorphic features including macrocephaly, hypertelorism, sparse eyebrows, broad forehead, prominent nose and pointed chin. The severity of the neurological effects as well as the presence of other features is variable among subjects. SNIBCPS is caused likely by pathogenic and pathogenic variants in CHD3 (Chromodomain Helicase DNA Binding Protein 3), which seems to be involved in chromatin remodeling by deacetylating histones. Here, we report 20 additional patients with clinical features compatible with SNIBCPS from 17 unrelated families with confirmed likely pathogenic/pathogenic variants in CHD3. Patients were analyzed by whole exome sequencing and segregation studies were performed by Sanger sequencing. Patients in this study showed different pathogenic variants affecting several functional domains of the protein. Additionally, none of the variants described here were reported in control population databases, and most computational predictors suggest that they are deleterious. The most common clinical features of the whole cohort of patients are global developmental delay (98%) and speech disorder/delay (92%). Other frequent features (51–74%) include intellectual disability, hypotonia, hypertelorism, abnormality of vision, macrocephaly and prominent forehead, among others. This study expands the number of individuals with confirmed SNIBCPS due to pathogenic or likely pathogenic variants in CHD3. Furthermore, we add evidence of the importance of the application of massive parallel sequencing for NDD patients for whom the clinical diagnosis might be challenging and where deep phenotyping is extremely useful to accurately manage and follow up the patients.
1. Introduction
Snijders Blok–Campeau syndrome (SNIBCPS) (MIM #618205) is a notably infrequent autosomal dominant disease, first described by Snijders Blok et al. in 2018 [1] and caused by pathogenic and likely pathogenic variants in the Chromodomain Helicase DNA Binding Protein 3 (CHD3) gene. The disorder shows an autosomal dominant pattern of inheritance. The most common clinical findings in affected patients include a neurodevelopmental disorder, characterized by global or speech delay, followed by either intellectual disability (ID) or specific learning difficulties, behavioral disorders like autism spectrum disorder (ASD) or attention deficit hyperactivity disorder (ADHD), and dysmorphic features including macrocephaly, prominent forehead, hypertelorism, face asymmetry, deeply set eyes, strabismus, epicanthus, wide nasal bridge, prominent nose, pointed chin, high palate and joint laxity among others [1]. The clinical features are highly heterogeneous and variable among individuals. Initially, 35 patients were described carrying a de novo pathogenic variant that disrupted the CHD3 gene [1]. To date, 63 patients have been diagnosed through molecular genetic techniques [1,2,3,4,5,6]. These patients had missense, in-frame deletions, nonsense and frameshift pathogenic or likely pathogenic variants. Additionally, one single case had a complete deletion of the CHD3 gene, and one patient was found to have a large duplication of the CHD3 gene [2].
Mammalian chromodomains helicase DNA-binding (CHDs) comprises a large family of proteins that have an indispensable role in developmental processes. Specifically, these proteins are involved in transcriptional regulation, and they can exert their chromatin remodeling activity to form the core ATPase subunit of the complex Nucleosome Remodeling Deacetylase (NuRD), which is associated with multiple cellular processes such as genomic integrity, cell cycle progression, or embryonic stem cell differentiation. Specifically, CHD3 encodes for a protein involved in late neural radial migration and cortical layer specification [2]. All these proteins have two chromodomains and two helicase domains. However, CHD3 additionally has two plant homeodomains (PHD), the function of which is to be responsible for the physical binding of CHDs to the NuRD chromatin-remodeling complex. Finally, it also contains domains of unknown function (DUF) [7] (Figure 1).
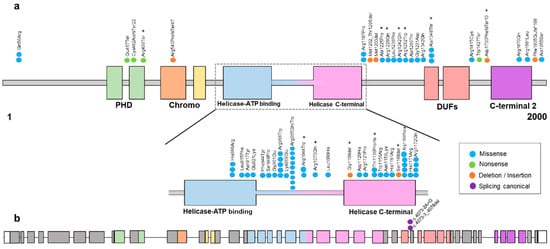
Figure 1.
(a) Schematic representation of the variants found in the CHD3 protein (transcript 1, NM_001005273.3) except for the splicing canonical variant that is shown in (b), found in our cohort and in the literature [1,2,3,4,5,6]. Most of the variants are found within the ATPase/helicase functional domain. The colors of missense, nonsense, deletion/insertion and splicing canonical are shown in the legend. * Variants reported in this study. (b) Schematic representation of the CHD3 exons (transcript 1, NM_001005273.3) with the splicing canonical variants.
In addition, according to previous findings and those described in this report, a variable degree of expression and reduced penetrance of the disease has been suggested [8]. Recently, it was observed that the majority of the variants that were inherited were maternally inherited. As mentioned by Spek et al. [8], the only parent in their cohort from whom a variant was inherited was affected, suggesting that female gender protects against genetic variation in disease.
Here, we describe 20 additional patients from 17 unrelated families with confirmed novel pathogenic variants in CHD3 and review the clinical and molecular characteristics of a total of 63 patients from the literature. Our work provides an updated clinical phenotype of patients with SNIBCPS and discusses the molecular pathogenesis and mechanism of inheritance. It confirms intrafamilial phenotypic variability. This article has special relevance for all professionals who are in charge of follow up patients with neurodevelopmental disorders (NDDs) and highlights the importance of an early diagnosis and the implementation of massive parallel sequencing technologies such as whole exome sequencing as the first tier for the diagnosis of these highly heterogeneous disorders.
2. Materials and Methods
2.1. Patient Cohort
All patients were recruited based upon either their clinical findings or the molecular defect in CHD3 from laboratories and hospitals in Spain, India, Italy and France. Informed consent was obtained from all patients and/or their legal guardians. Neuropsychiatric findings were evaluated by a specialist for each patient. Some of the patients from Hospital Universitario La Paz are part of the Spanish Overgrowth Registry Initiative (SOGRI) database.
2.2. Genetic Analysis
Patients were mostly analyzed by whole exome sequencing (WES). Segregation analysis was performed in parental samples by Sanger sequencing when samples were available. Variants are described using the HGVS nomenclature and classified according to the ACMG guidelines [9]. The in silico pathogenicity of each of the variants was assessed using CADD-PHRED v1.6 and REVEL v4.3 scores.
3. Results
By means of the application of massive parallel sequencing technologies, as well as international collaboration through scientific networks, we have identified 20 additional cases with SNIBCPS with pathogenic or likely pathogenic variants in CHD3. Segregation analysis showed that variants were de novo in 16 out of 20 patients (80%), inherited in 3 others (15%), and segregation analysis was not possible for one of them.
At the clinical level, all 20 patients were phenotyped, and the identified clinical features are summarized in Table 1. The most frequent (>75%) clinical features observed in these patients included global developmental delay and speech disorder/delay. Although global developmental delay was almost constant, including generalized hypotonia and language delay, almost half of the patients aged 5 years old and beyond showed no ID. Two of our patients presented with ASD, and six of them with ADHD.

Table 1.
Clinical features found in patients with SNIBCPS. HPO prioritization frequency of clinical features described in patients with CHD3 variants. Percentage was calculated according to the total number of patients reported in the literature. Patients compiled from [1,2,3,4,5,6] (63 patients) and the 20 patients reported herein.
In addition, in our cohort we present a family (P12, 13 and 14, Table 2) in which mother and two children have variants in the CHD3 gene. Both mother and daughter presented with initial global developmental delay but had no ID and a diagnosis of developmental coordination disorder. The son presented with ASD and moderate ID.
Table 2.
Variants detected in CHD3 in our cohort. ACMG, American College of Medical Genetics. # Population frequency was estimated from pseudo-control databases: GnomAD genomes (v3.0); GnomAD exomes (v3.1); Kaviar (version160204-Public); Beacon (v2.0); 1000 G; Phase III; and Bravo (TOVMed Freeze 8).
Other frequent clinical features (50–75%) observed were: intellectual disability, hypotonia, hypertelorism, abnormality of vision, macrocephaly and prominent forehead.
At the molecular level, 16 out the 20 variants detected were missense and the other four included a nonsense, a frameshift deletion, and two in-frame deletions. All variants were heterozygous.
The majority of the detected variants were located within or between the helicase ATP-binding domain, the helicase C-terminal domain and the DUF domain except for the frameshift variant, which is located before the C-terminal 2 domain and the transcript encoded is predicted to be degraded by the nonsense-mediated decay (NMD) mechanism (Figure 1).
After ACMG variant classification, eight variants were classified as pathogenic and 12 as likely pathogenic. None of the variants reported herein were previously reported and/or associated with SNIBCPS. Moreover, none of the variants described in our series have previously been reported in pseudo-control population databases (gnomAD exomes, gnomAD genomes, Kaviar, Beacon, 1000G, ESP and Bravo) and the majority of the computational evidence support a deleterious/pathogenic effect for all the variants reported in this study (Table 2).
4. Discussion
NDDs comprise a large and highly heterogeneous group of disorders, caused by a variety of pathogenic and likely pathogenic variants in >1000 genes and in which there might by a highly clinical presentation overlap. With the advances in massive parallel sequencing technologies, it has become possible to identify and confirm or discard initial clinical suspicions in this group of patients.
SNIBCPS was first described in 2018 by Snijders Blok et al. [1]. SNIBCPS can be classified as an NDD, and because it is caused by pathogenic variants in CHD3, it may have phenotypic overlap with other human diseases caused by mutations in CHD proteins. Some of these may include CHD7 causing CHARGE syndrome (MIM #214800), CHD4 as causative of Sifrim–Hitz–Weiss syndrome (MIM #603277), or CHD8 causing autism spectrum disorder (MIM #610528).
One of the challenges in the recognition of the SNIBCPS syndrome is the highly phenotypic overlap with other genetic disorders, especially NDDs and overgrowth syndromes, because the most common findings are macrocephaly and neurodevelopmental problems. Besides NDDs and enlarged head circumference, some dysmorphic features such as pointed chin or frontal bossing may resemble syndromes such as Sotos, Malan, Tenorio, or Simpson–Golabi–Behmel syndromes [10,11,12,13].
NuRD is an ATP-dependent chromatin remodeling complex. It consists of multiple proteins, including CHD3 and CHD4, which are important for the activity of the NuRD complex, as they provide the energy and helicase function necessary for chromatin remodeling. This complex has important functions in cellular processes such as peripheral nerve myelination and cortical development in the brain. CHD3 is an ATP-dependent chromatin remodeling protein that serves as core member of the NuRD complex [14] and it has been shown to play an important role in the viability of the developing embryonic brain [15].
In this study, we report 20 patients from 17 unrelated families with a clinical phenotype compatible with SNIBCPS in whom a pathogenic/likely pathogenic variant in the CHD3 gene was identified. Thus, thanks to the increase in the number of patients with CHD3 defects, it was possible to expand the SNIBCPS phenotype.
Reviewing our cohort of patients, we observed that global developmental delay and speech disorder/delay are the most common (>75%) clinical features. The clinical characteristics observed in 50–75% of individuals were intellectual disability, hypotonia, hypertelorism, abnormality of vision, macrocephaly and prominent forehead. Less common clinical features (25–50%) were abnormality of CNS, autism and some dysmorphic features such as pointed chin or wide nasal bridge, among others (Table 1). In addition, we were able to expand the phenotypic spectrum with other unreported features such as epicanthus (12%), attention deficit hyperactivity disorder (ADHD) (7%), high palate (6%), foot deformities (5%), depressed nasal root (5%) and blushed cheeks (4%). Our series confirms intrafamilial clinical variability highlighting the importance of paying attention to the variants transmitted when interpreting WES or WGS analyses.
We found 20 likely pathogenic/pathogenic variants. On the other hand, it has previously been reported that most of the variants described are located within the ATPase/helicase domain of the CHD3 gene [2].
Of the 20 individuals in our cohort, eight of them had pathogenic variants in the ATPase/helicase domain, with 12 variants being outside this domain. It has previously been reported that there are no phenotypic differences between patients with variants within the ATPase/helicase domain and individuals with variants outside this domain [2].
In our series, we also found no phenotypic differences between patients with a variant within the ATPase/helicase domain and those with a variant outside of this domain.
CHD3 is extremely intolerant to LoF (loss-of-function) and missense variation based on in silico analysis (probability of LoF intolerance = 1, observed/expected = 0.09 [0.05–0.15]; Z-score = 6.15, observed/expected = 0.5 [0.46–0.53]). These values suggest that a haploinsufficiency mechanism may be the cause of the disease, as other studies have already suggested [8]. We analyzed the distribution of CCRs (Constrained Coding Regions) and found that four of the variants present in our cohort of patients had highly constrained regions with percentiles in the interval (90–100).
In addition, previous studies have sought to determine the effect of variants in CHD3 using an ATPase assay. A clear decrease in ATPase activity was observed in two variants (p.Arg1121Pro and p.Arg1172Gln) [1]. As mentioned above, in our study, 13 variants were found in the ATPase functional domain, half of them in the amino acid arginine (Figure 1). Moreover, it was observed that ATP binds strongly to Arg and with high affinity, and Arg dominates the direct binding of ATP and affects the self-association of accumulated ATPs. The size of the ATP pool is effectively regulated by the distribution of Arg [16]. Our hypothesis regarding these findings is that the effect of these variants on CHD3 (p.Arg985Trp, p.Arg1044Trp, p.Arg1070Gln and p.Arg1169Trp) may affect critical functions of the protein, such as impacting the ability of the protein to interact with other molecules or proteins involved in chromatin remodeling processes. On the other hand, it can influence the stability and folding capacity of the protein to adopt its three-dimensional structure, leading to protein misfolding and/or instability. In order to fully understand the consequences of the different variants in CHD3, further functional analysis and protein modeling techniques are needed.
In our series, segregation analysis showed that three of the patients had a variant inherited from one of their parents (15%). In one of these families, the transmitting parent was apparently healthy. It has been reported that most of the variants inherited in other cohorts were maternally inherited, suggesting that female gender protects against genetic variations in this disease. This observation may indicate incomplete penetrance and variable expressivity in this syndrome [8].
In conclusion, we herein report 20 additional patients with novel variants along the CHD3 gene. The main clinical manifestations are the presence of neurodevelopmental problems without intellectual disability in half of the patients as well as dysmorphic features, such as macrocephaly. At the molecular level, we found in our cohort, as well as in the cohorts described above, that most of the variants are located in the ATPase/helicase functional domain. Thus, we have increased the number of patients with SNIBCPS as well as expanded the phenotypic features of this unusual disorder. Further studies may help to elucidate genotype–phenotype correlations and to understand the mechanism by which a CHD3 gene defect causes Snijders Blok–Campeau syndrome (OMIM #518205).
Author Contributions
Conceptualization, P.L. and J.T.-C.; methodology, P.P., P.A., C.S.; software, P.P., A.P., L.M., M.C., N.G.-Z.; formal analysis, P.P., J.T.-C., C.M., A.A.; investigation, P.P., P.L., J.T.-C.; resources, D.H., C.M., A.A., B.K., I.P., M.P., E.R., F.J.R., B.A., C.A., S.T.S., A.I.S.B., V.M.S., V.K.G., M.M., V.N., S.D., C.C., C.M.M., B.P., G.G., A.T.S.A., M.D.J., V.S., J.S., V.C.-D., The SOGRI Consortium, J.N.; data curation, P.P., J.N.; writing—original draft preparation, P.P., J.T.-C., P.L.; writing—review and editing, J.T.-C., P.L., J.N.; supervision, P.L., J.T.-C.; funding acquisition, J.T.-C., P.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by Telethon Undiagnosed Diseases Program (TUDP, GSP15001), ISCIII-Feder funds: grants FIS PI20/01053, PMP22/00049.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of La Paz University Hospital (protocol code PI-5534 date of approval 31/01/2023).” for studies involving humans.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available in tables and figures of the article.
Acknowledgments
We would like to thank all the families who were involved in this project, as well as the participants of the Spanish OverGrowth Registry Initiative (SOGRI) Consortium. The SOGRI (Spanish OverGrowth Registry Initiative) Consortium is comprised of the following researchers: Antonio Plasencia, Alberto L. Rosa, Aleixandre Blanquer, Alfredo Garcı’a-Alix, Alfredo Santana, Alicia Delicado, Almudena Alonso, Amaya Rodriguez, Amparo Sanchis, Ana Moreno, Ana Patiño García, Ana Vega, Analía Bredani, Andrea Paula Solari, Andrea Villavicencio, Angelina Acosta, Anibal Nieto, Anna María Cueto González, Antonio Baldellón, Antonio González Meneses, Antonio Martínez Carrascal, Aranzazu Díaz de Bustamante, Arteche Ocasar, Blanca Gener, Blasco González, Boris Groisman, Bradford Coffee, Carlos Alcalde Martín, Carmen Aragon Fernández, Carmen Benito, Carmen Martin Seisdedos, Carmen Roche, Claudia Arberas, Claudia Perandones, Claudio Contessotto, Cristina Olivas, Daniel Armenta, Denise Cavalcanti, Dolores Elorza, Elena Zamora, Elisa Zambrano, Elisabeth Steichen, Enrique Caro Cruz, Enrique Galán Gómez, Enriqueta Román, Ernesto Goldschmidt, Esteban Marfil, Esther Gean, Eugenia Antolín, F. Javier Gascón Jiménez, Feliciano Ramos, Fermina López Grondona, Fernández Córdoba, Fernando Regla Vargas, Francisco Martínez, J. Miguel García Vegada, Giovannucci Uzielli, Gloria Gacio, Carmen González Armengod, Graciela Mercado, Hamilton Cassinelli, Ieda Orioli, Ignacio Arroyo, Ignacio Díez López, Ignacio Onsurbe Ramírez, Ignacio Pascual Castroviejo, Ignacio Pascual Pascual, Ignacio Vázquez Rio, Inés Bueno, Isabel Espejo Portero, Isabel Lorda Sánchez, Jaime Sánchez del Pozo, Jaume Campistol, Javier Arcas, Javier Fernández, Javier García Planells, Javier López Pisón, Jesús Barreiro, Jesús del Valle Nuñez, María José Jiménez Liria, Joaquín Fernández Toral, Joaquín Ramírez, Jordi Rosell, Jorge Vilaplana, José Carlos Cabral de Almeida, José Ignacio Labarta, José L. Herranz, José Luis Fernández Luna, José Luis Fuster, José M. Díaz, Jose M. Gairi, José Miguel García Sagredo, Juan A. Piñero, Juan Carlos López Gutiérrez, Juan Manuel Fernández, Juan P. López Siguero, JuanTovar, Judith Armstrong, Julián Lara, Leonor Arranz, Laura Rodríguez, Leandro Soriano, Liliana De Alba, Loreta Cimbalistiene, Loreto Martorell, Luis González Gutiérrez Solana, Luis Pérez Jurado, M Asunción López Ariztegui, M. Antonia Molina, M. Cruz García, M. Ferrer Lozano, M. Jesús Alija Merillas, M. Luisa Martínez-Frías, María L. Martínez Fernández, M. Rocío Jadraque, María Asunción García Pérez, María Montserrat Rodríguez Pedreira, María Pilar Ribate, María Teresa González López, María Teresa Moral Pumarega, Mabel Segovia, Macarena Lizama, Manuel Pombo, Margarita Martínez, Margarita Tabernero, María Antonia Ramos, Maria Ballesta, María Belar, María Jesús Lautre, Marta Cruz, M. Nieves Martínez Guardia, F. Javier Martínez Sarries, Mercedes Artigas, Mercedes Villanueva, Meritxell Torrabías, Miguel del Campo, Miguel Tomás Vila, Miguel Urioste, Mónica Rosello, Nik Kantaputra, Pablo Prieto Matos, Paloma Dorao, Paula Casano, Paula Lalaguna Mallada, Pedro Olivares, Raquel Perez Delgado, Priscila Bernardi, Rafael Camino León, Ramón Cañete, Ramón Gaztañaga, Ramón Velazquez, Ramón Vidal Samahuja, Raquel Sáez Villaverde, Ricardo Gracia, Richard Scott, Rita Valdez, Rosa Arteaga, Rosa Cedeño, Rosario Cazorla, Rosario Marín Iglesias, Rubén Bronberg, Salvador Climent, Santiago Conde Barreiro, Seema Kapoor, Soledad Kleppe, Sonia Santillán, Trinidad García Lopez, Teresa Calvo, Teresa Vendrell, Pilar Tirado, Claudia Toledo Pacheco, Alicia Ureta Huertos, Vanesa Lopez, Vanesa Lotersztein, Vanesa Méndez, Selma Vázquez Martín, Verónica Seidel, Vicente Albiach, Víctor M. Navas López, Virgina Soler, and Viviana Cosentino.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Snijders Blok, L.; Rousseau, J.; Twist, J.; Ehresmann, S.; Takaku, M.; Venselaar, H.; Rodan, L.H.; Nowak, C.B.; Douglas, J.; Swoboda, K.J.; et al. CHD3 Helicase Domain Mutations Cause a Neurodevelopmental Syndrome with Macrocephaly and Impaired Speech and Language. Nat. Commun. 2018, 9, 4619. [Google Scholar] [CrossRef] [PubMed]
- Drivas, T.G.; Li, D.; Nair, D.; Alaimo, J.T.; Alders, M.; Altmüller, J.; Barakat, T.S.; Bebin, E.M.; Bertsch, N.L.; Blackburn, P.R.; et al. A Second Cohort of CHD3 Patients Expands the Molecular Mechanisms Known to Cause Snijders Blok-Campeau Syndrome. Eur. J. Hum. Genet. 2020, 28, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Coursimault, J.; Lecoquierre, F.; Saugier-Veber, P.; Drouin-Garraud, V.; Lechevallier, J.; Boland, A.; Deleuze, J.-F.; Frebourg, T.; Nicolas, G.; Brehin, A.-C. Hypersociability Associated with Developmental Delay, Macrocephaly and Facial Dysmorphism Points to CHD3 Mutations. Eur. J. Med. Genet. 2021, 64, 104166. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, M.; Ishikawa, A.; Miyazaki, S.; Tsuzuki, A.; Saito, S.; Niihori, T.; Sakurai, A. A de Novo CHD3 Variant in a Child with Intellectual Disability, Autism, Joint Laxity, and Dysmorphisms. Brain Dev. 2021, 43, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.-Y. Snijders Blok-Campeau Syndrome Caused by CHD3 Gene Mutation: A Case Report. Zhongguo Dang Dai Er Ke Za Zhi Chin. J. Contemp. Pediatr. 2021, 23, 965–968. [Google Scholar]
- LeBreton, L.; Allain, E.P.; Parscan, R.C.; Crapoulet, N.; Almaghraby, A.; Ben Amor, M. A Novel CHD3 Variant in a Patient with Central Precocious Puberty: Expanded Phenotype of Snijders Blok-Campeau Syndrome? Am. J. Med. Genet. A 2023, 191, 1065–1069. [Google Scholar] [CrossRef]
- Cardoso, A.R.; Lopes-Marques, M.; Oliveira, M.; Amorim, A.; Prata, M.J.; Azevedo, L. Genetic Variability of the Functional Domains of Chromodomains Helicase DNA-Binding (CHD) Proteins. Genes 2021, 12, 1827. [Google Scholar] [CrossRef] [PubMed]
- Van der Spek, J.; den Hoed, J.; Snijders Blok, L.; Dingemans, A.J.M.; Schijven, D.; Nellaker, C.; Venselaar, H.; Astuti, G.D.N.; Barakat, T.S.; Bebin, E.M.; et al. Inherited Variants in CHD3 Show Variable Expressivity in Snijders Blok-Campeau Syndrome. Genet. Med. 2022, 24, 1283–1296. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Rahman, N. Sotos Syndrome. Eur. J. Hum. Genet. 2007, 15, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Priolo, M.; Schanze, D.; Tatton-Brown, K.; Mulder, P.A.; Tenorio, J.; Kooblall, K.; Acero, I.H.; Alkuraya, F.S.; Arias, P.; Bernardini, L.; et al. Further Delineation of Malan Syndrome. Hum. Mutat. 2018, 39, 1226–1237. [Google Scholar] [CrossRef]
- Tenorio, J.; Mansilla, A.; Valencia, M.; Martínez-Glez, V.; Romanelli, V.; Arias, P.; Castrejón, N.; Poletta, F.; Guillén-Navarro, E.; Gordo, G.; et al. A New Overgrowth Syndrome Is Due to Mutations in RNF125. Hum. Mutat. 2014, 35, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Tenorio, J.; Arias, P.; Martínez-Glez, V.; Santos, F.; García-Miñaur, S.; Nevado, J.; Lapunzina, P. Simpson-Golabi-Behmel Syndrome Types I and II. Orphanet J. Rare Dis. 2014, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Torchy, M.P.; Hamiche, A.; Klaholz, B.P. Structure and Function Insights into the NuRD Chromatin Remodeling Complex. Cell. Mol. Life Sci. 2015, 72, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Gao, S.; Schafer, C.; Colijn, S.; Muthukumar, V.; Griffin, C.T. The Chromatin-Remodeling Enzyme CHD3 Plays a Role in Embryonic Viability but Is Dispensable for Early Vascular Development. PLoS ONE 2020, 15, e0235799. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Ou, X.; Li, J. Mechanistic Insight on General Protein-Binding Ability of ATP and the Impacts of Arginine Residues. J. Phys. Chem. B 2022, 126, 4647–4658. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).