
Integrative Analysis Unveils the Correlation of Aminoacyl-tRNA Biosynthesis Metabolites with the Methylation of the SEPSECS Gene in Huntington’s Disease Brain Tissue
 ,
,  , and
, and 
Abstract
:1. Introduction
2. Methods
2.1. Study Samples
2.2. 1H NMR Analysis
2.3. Direct Injection/Liquid Chromatography–Mass Spectral Analysis (DI/LC-MS/MS)
2.4. Genome-Wide DNA Methylation Assay
2.5. Data Analysis
2.6. Epigenome–Metabolome Interactions
2.7. Diagnostic Models
3. Results
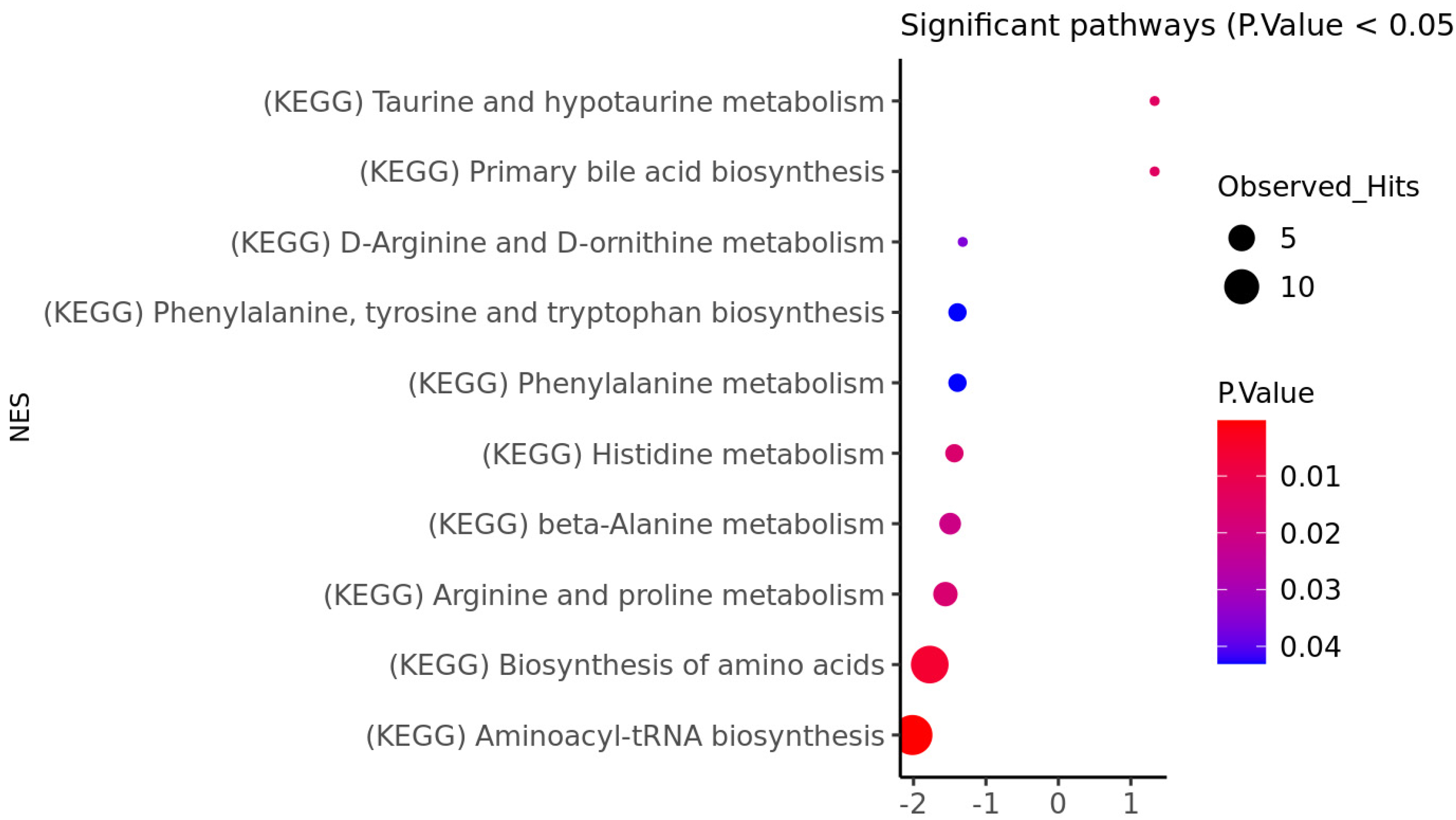
3.1. HD Brain Metabolomic Profile
3.2. HD Brain Methylation Profile
3.3. Epigenome and Metabolome Correlation
3.4. Diagnostic Models
4. Discussion
Aminoacyl-tRNA Biosynthesis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stoker, T.B.; Mason, S.L.; Greenland, J.C.; Holden, S.T.; Santini, H.; Barker, R.A. Huntington’s disease: Diagnosis and management. Pract. Neurol. 2022, 22, 32–41. [Google Scholar] [CrossRef]
- Finkbeiner, S. Huntington’s Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a007476. [Google Scholar] [CrossRef] [PubMed]
- Roos, R.A. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef]
- Hashimoto, M.; Watanabe, K.; Miyoshi, K.; Koyanagi, Y.; Tadano, J.; Miyawaki, I. Multiplatform metabolomic analysis of the R6/2 mouse model of Huntington’s disease. FEBS Open Bio 2021, 11, 2807–2818. [Google Scholar] [CrossRef] [PubMed]
- Medina, A.; Mahjoub, Y.; Shaver, L.; Pringsheim, T. Prevalence and Incidence of Huntington’s Disease: An Updated Systematic Review and Meta-Analysis. Mov. Disord. Off. J. Mov. Disord. Soc. 2022, 37, 2327–2335. [Google Scholar] [CrossRef]
- Scahill, R.I.; Zeun, P.; Osborne-Crowley, K.; Johnson, E.B.; Gregory, S.; Parker, C.; Lowe, J.; Nair, A.; O’Callaghan, C.; Langley, C.; et al. Biological and clinical characteristics of gene carriers far from predicted onset in the Huntington’s disease Young Adult Study (HD-YAS): A cross-sectional analysis. Lancet Neurol. 2020, 19, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Blumenstock, S.; Dudanova, I. Cortical and Striatal Circuits in Huntington’s Disease. Front. Neurosci 2020, 14, 82. [Google Scholar] [CrossRef]
- Waldvogel, H.J.; Kim, E.H.; Tippett, L.J.; Vonsattel, J.P.; Faull, R.L. The Neuropathology of Huntington’s Disease. Curr. Top. Behav. Neurosci. 2015, 22, 33–80. [Google Scholar] [CrossRef]
- Gatto, E.M.; Rojas, N.G.; Persi, G.; Etcheverry, J.L.; Cesarini, M.E.; Perandones, C. Huntington disease: Advances in the understanding of its mechanisms. Clin. Park. Relat. Disord. 2020, 3, 100056. [Google Scholar] [CrossRef]
- Chow, T.W. Personality in frontal lobe disorders. Curr. Psychiatry Rep. 2000, 2, 446–451. [Google Scholar] [CrossRef]
- Duff, K.; Paulsen, J.S.; Beglinger, L.J.; Langbehn, D.R.; Wang, C.; Stout, J.C.; Ross, C.A.; Aylward, E.; Carlozzi, N.E.; Queller, S. “Frontal” behaviors before the diagnosis of Huntington’s disease and their relationship to markers of disease progression: Evidence of early lack of awareness. J. Neuropsychiatry Clin. Neurosci. 2010, 22, 196–207. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P., Jr. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Aylward, E.H.; Anderson, N.B.; Bylsma, F.W.; Wagster, M.V.; Barta, P.E.; Sherr, M.; Feeney, J.; Davis, A.; Rosenblatt, A.; Pearlson, G.D.; et al. Frontal lobe volume in patients with Huntington’s disease. Neurology 1998, 50, 252–258. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef]
- Villa, C.; Yoon, J.H. Multi-Omics for the Understanding of Brain Diseases. Life 2021, 11, 1202. [Google Scholar] [CrossRef]
- Paananen, J.; Fortino, V. An omics perspective on drug target discovery platforms. Brief Bioinform. 2020, 21, 1937–1953. [Google Scholar] [CrossRef]
- Lu, H.; Liu, X.; Deng, Y.; Qing, H. DNA methylation, a hand behind neurodegenerative diseases. Front. Aging Neurosci. 2013, 5, 85. [Google Scholar] [CrossRef]
- Zsindely, N.; Siági, F.; Bodai, L. DNA Methylation in Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 12736. [Google Scholar] [CrossRef]
- Lu, A.T.; Narayan, P.; Grant, M.J.; Langfelder, P.; Wang, N.; Kwak, S.; Wilkinson, H.; Chen, R.Z.; Chen, J.; Simon Bawden, C.; et al. DNA methylation study of Huntington’s disease and motor progression in patients and in animal models. Nat. Commun. 2020, 11, 4529. [Google Scholar] [CrossRef]
- Pan, Y.; Daito, T.; Sasaki, Y.; Chung, Y.H.; Xing, X.; Pondugula, S.; Swamidass, S.J.; Wang, T.; Kim, A.H.; Yano, H. Erratum: Inhibition of DNA Methyltransferases Blocks Mutant Huntingtin-Induced Neurotoxicity. Sci. Rep. 2016, 6, 33766. [Google Scholar] [CrossRef]
- Feehley, T.; O’Donnell, C.W.; Mendlein, J.; Karande, M.; McCauley, T. Drugging the epigenome in the age of precision medicine. Clin. Epigenetics 2023, 15, 6. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Covarrubias, V.; Martinez-Martinez, E.; Del Bosque-Plata, L. The Potential of Metabolomics in Biomedical Applications. Metabolites 2022, 12, 194. [Google Scholar] [CrossRef] [PubMed]
- Skene, D.J.; Middleton, B.; Fraser, C.K.; Pennings, J.L.; Kuchel, T.R.; Rudiger, S.R.; Bawden, C.S.; Morton, A.J. Metabolic profiling of presymptomatic Huntington’s disease sheep reveals novel biomarkers. Sci. Rep. 2017, 7, 43030. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sun, H.; Yan, G.; Wang, P.; Wang, X. Metabolomics for Biomarker Discovery: Moving to the Clinic. Biomed Res. Int. 2015, 2015, 354671. [Google Scholar] [CrossRef]
- Rosas, H.D.; Doros, G.; Bhasin, S.; Thomas, B.; Gevorkian, S.; Malarick, K.; Matson, W.; Hersch, S.M. A systems-level “misunderstanding”: The plasma metabolome in Huntington’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 756–768. [Google Scholar] [CrossRef]
- Underwood, B.R.; Broadhurst, D.; Dunn, W.B.; Ellis, D.I.; Michell, A.W.; Vacher, C.; Mosedale, D.E.; Kell, D.B.; Barker, R.A.; Grainger, D.J.; et al. Huntington disease patients and transgenic mice have similar pro-catabolic serum metabolite profiles. Brain 2006, 129, 877–886. [Google Scholar] [CrossRef]
- Leoni, V.; Caccia, C. The impairment of cholesterol metabolism in Huntington disease. Biochim. Biophys. Acta 2015, 1851, 1095–1105. [Google Scholar] [CrossRef]
- Block, R.C.; Dorsey, E.R.; Beck, C.A.; Brenna, J.T.; Shoulson, I. Altered cholesterol and fatty acid metabolism in Huntington disease. J. Clin. Lipidol. 2010, 4, 17–23. [Google Scholar] [CrossRef]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Transcriptional control of amino acid homeostasis is disrupted in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2016, 113, 8843–8848. [Google Scholar] [CrossRef]
- Graham, S.F.; Pan, X.; Yilmaz, A.; Macias, S.; Robinson, A.; Mann, D.; Green, B.D. Targeted biochemical profiling of brain from Huntington’s disease patients reveals novel metabolic pathways of interest. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 2430–2437. [Google Scholar] [CrossRef]
- Graham, S.F.; Kumar, P.K.; Bjorndahl, T.; Han, B.; Yilmaz, A.; Sherman, E.; Bahado-Singh, R.O.; Wishart, D.; Mann, D.; Green, B.D. Metabolic signatures of Huntington’s disease (HD): (1)H NMR analysis of the polar metabolome in post-mortem human brain. Biochim. Biophys. Acta 2016, 1862, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Mercier, P.; Lewis, M.J.; Chang, D.; Baker, D.; Wishart, D.S. Towards automatic metabolomic profiling of high-resolution one-dimensional proton NMR spectra. J. Biomol. NMR 2011, 49, 307–323. [Google Scholar] [CrossRef]
- Urban, M.; Enot, D.P.; Dallmann, G.; Körner, L.; Forcher, V.; Enoh, P.; Koal, T.; Keller, M.; Deigner, H.-P. Complexity and pitfalls of mass spectrometry-based targeted metabolomics in brain research. Anal. Biochem. 2010, 406, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.F.; Turkoglu, O.; Kumar, P.; Yilmaz, A.; Bjorndahl, T.C.; Han, B.; Mandal, R.; Wishart, D.S.; Bahado-Singh, R.O. Targeted Metabolic Profiling of Post-Mortem Brain from Infants Who Died from Sudden Infant Death Syndrome. J. Proteome Res. 2017, 16, 2587–2596. [Google Scholar] [CrossRef]
- Graham, S.F.; Chevallier, O.P.; Kumar, P.; Türko Gcaron Lu, O.; Bahado-Singh, R.O. Metabolomic profiling of brain from infants who died from Sudden Infant Death Syndrome reveals novel predictive biomarkers. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2017, 37, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.A. DNA methylation in Huntington’s disease: Implications for transgenerational effects. Neurosci. Lett. 2016, 625, 34–39. [Google Scholar] [CrossRef]
- Katada, S.; Imhof, A.; Sassone-Corsi, P. Connecting Threads: Epigenetics and Metabolism. Cell 2012, 148, 24–28. [Google Scholar] [CrossRef]
- Kori, M.; Aydın, B.; Unal, S.; Arga, K.Y.; Kazan, D. Metabolic Biomarkers and Neurodegeneration: A Pathway Enrichment Analysis of Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis. OMICS A J. Integr. Biol. 2016, 20, 645–661. [Google Scholar] [CrossRef]
- Herman, S.; Niemelä, V.; Emami Khoonsari, P.; Sundblom, J.; Burman, J.; Landtblom, A.-M.; Spjuth, O.; Nyholm, D.; Kultima, K. Alterations in the tyrosine and phenylalanine pathways revealed by biochemical profiling in cerebrospinal fluid of Huntington’s disease subjects. Sci. Rep. 2019, 9, 4129. [Google Scholar] [CrossRef] [PubMed]
- Young, S.N.; Shalchi, M. The effect of methionine and S-adenosylmethionine on S-adenosylmethionine levels in the rat brain. J. Psychiatry Neurosci. JPN 2005, 30, 44–48. [Google Scholar]
- Tapia-Rojas, C.; Lindsay, C.B.; Montecinos-Oliva, C.; Arrazola, M.S.; Retamales, R.M.; Bunout, D.; Hirsch, S.; Inestrosa, N.C. Is L-methionine a trigger factor for Alzheimer’s-like neurodegeneration?: Changes in Aβ oligomers, tau phosphorylation, synaptic proteins, Wnt signaling and behavioral impairment in wild-type mice. Mol. Neurodegener. 2015, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- Schöneich, C. Methionine oxidation by reactive oxygen species: Reaction mechanisms and relevance to Alzheimer’s disease. Biochim. Biophys. Acta 2005, 1703, 111–119. [Google Scholar] [CrossRef]
- Selhub, J. Homocysteine metabolism. Annu. Rev. Nutr. 1999, 19, 217–246. [Google Scholar] [CrossRef] [PubMed]
- Andrich, J.; Saft, C.; Arz, A.; Schneider, B.; Agelink, M.W.; Kraus, P.H.; Kuhn, W.; Müller, T. Hyperhomocysteinaemia in treated patients with Huntington’s disease homocysteine in HD. Mov. Disord. Off. J. Mov. Disord. Soc. 2004, 19, 226–228. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Gao, M.; Su, Y.; Liu, P.; Sun, B. Running Promotes Transformation of Brain Astrocytes Into Neuroprotective Reactive Astrocytes and Synaptic Formation by Targeting Gpc6 Through the STAT3 Pathway. Front. Physiol. 2021, 12, 633618. [Google Scholar] [CrossRef] [PubMed]
- Baier, A.; Szyszka, R. CK2 and protein kinases of the CK1 superfamily as targets for neurodegenerative disorders. Front. Mol. Biosci. 2022, 9, 916063. [Google Scholar] [CrossRef]
- Geisinger, A.; Alsheimer, M.; Baier, A.; Benavente, R.; Wettstein, R. The mammalian gene pecanex 1 is differentially expressed during spermatogenesis. Biochim. Biophys. Acta 2005, 1728, 34–43. [Google Scholar] [CrossRef]
- Song, W.; Li, Q.; Wang, T.; Li, Y.; Fan, T.; Zhang, J.; Wang, Q.; Pan, J.; Dong, Q.; Sun, Z.S.; et al. Putative complement control protein CSMD3 dysfunction impairs synaptogenesis and induces neurodevelopmental disorders. Brain Behav. Immun. 2022, 102, 237–250. [Google Scholar] [CrossRef]
- Xu, A.; Wang, W.; Jiang, X. The roles of MTRR and MTHFR gene polymorphisms in congenital heart diseases: A meta-analysis. Biosci. Rep. 2018, 38, BSR20181160. [Google Scholar] [CrossRef]
- Wang, Y.; Kerrisk Campbell, M.; Tom, I.; Foreman, O.; Hanson, J.E.; Sheng, M. PCDH7 interacts with GluN1 and regulates dendritic spine morphology and synaptic function. Sci. Rep. 2020, 10, 10951. [Google Scholar] [CrossRef]
- Yamada, S.B.; Gendron, T.F.; Niccoli, T.; Genuth, N.R.; Grosely, R.; Shi, Y.; Glaria, I.; Kramer, N.J.; Nakayama, L.; Fang, S.; et al. RPS25 is required for efficient RAN translation of C9orf72 and other neurodegenerative disease-associated nucleotide repeats. Nat. Neurosci. 2019, 22, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Wild, E.J.; Tabrizi, S.J. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017, 16, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.G.; Scala, M.; Beetz, C.; Helman, G.; Stanley, V.; Yang, X.; Breuss, M.W.; Mazaheri, N.; Selim, L.; Hadipour, F.; et al. A relatively common homozygous TRAPPC4 splicing variant is associated with an early-infantile neurodegenerative syndrome. Eur. J. Hum. Genet. EJHG 2021, 29, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Rubio Gomez, M.A.; Ibba, M. Aminoacyl-tRNA synthetases. RNA 2020, 26, 910–936. [Google Scholar] [CrossRef]
- Ibba, M.; Söll, D. Aminoacyl-tRNAs: Setting the limits of the genetic code. Genes Dev. 2004, 18, 731–738. [Google Scholar] [CrossRef]
- Girstmair, H.; Saffert, P.; Rode, S.; Czech, A.; Holland, G.; Bannert, N.; Ignatova, Z. Depletion of cognate charged transfer RNA causes translational frameshifting within the expanded CAG stretch in huntingtin. Cell Rep. 2013, 3, 148–159. [Google Scholar] [CrossRef]
- Lant, J.T.; Berg, M.D.; Heinemann, I.U.; Brandl, C.J.; O’Donoghue, P. Pathways to disease from natural variations in human cytoplasmic tRNAs. J. Biol. Chem. 2019, 294, 5294–5308. [Google Scholar] [CrossRef]
- Lant, J.T.; Kiri, R.; Duennwald, M.L.; O’Donoghue, P. Formation and persistence of polyglutamine aggregates in mistranslating cells. Nucleic Acids Res. 2021, 49, 11883–11899. [Google Scholar] [CrossRef]
- Mochel, F.; Charles, P.; Seguin, F.; Barritault, J.; Coussieu, C.; Perin, L.; Le Bouc, Y.; Gervais, C.; Carcelain, G.; Vassault, A.; et al. Early energy deficit in Huntington disease: Identification of a plasma biomarker traceable during disease progression. PLoS ONE 2007, 2, e647. [Google Scholar] [CrossRef]
- Schmidt, R.L.; Simonović, M. Synthesis and decoding of selenocysteine and human health. Croat. Med. J. 2012, 53, 535–550. [Google Scholar] [CrossRef]
- Zhang, Y.; Roh, Y.J.; Han, S.J.; Park, I.; Lee, H.M.; Ok, Y.S.; Lee, B.C.; Lee, S.R. Role of Selenoproteins in Redox Regulation of Signaling and the Antioxidant System: A Review. Antioxidants 2020, 9, 383. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ratan, R.R. Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly. J. Huntington’s Dis. 2016, 5, 217–237. [Google Scholar] [CrossRef]
- Browne, S.E.; Beal, M.F. Oxidative damage in Huntington’s disease pathogenesis. Antioxid. Redox Signal. 2006, 8, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Machiela, E.; Jeloka, R.; Caron, N.S.; Mehta, S.; Schmidt, M.E.; Baddeley, H.J.E.; Tom, C.M.; Polturi, N.; Xie, Y.; Mattis, V.B.; et al. The Interaction of Aging and Cellular Stress Contributes to Pathogenesis in Mouse and Human Huntington Disease Neurons. Front. Aging Neurosci. 2020, 12, 524369. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.; Uyehara-Lock, J.H.; Bellinger, F.P. Selenium and selenoprotein function in brain disorders. IUBMB Life 2014, 66, 229–239. [Google Scholar] [CrossRef]
- Cardoso, B.R.; Roberts, B.R.; Bush, A.I.; Hare, D.J. Selenium, selenoproteins and neurodegenerative diseases. Met. Integr. Biometal Sci. 2015, 7, 1213–1228. [Google Scholar] [CrossRef]
- Umair, M.; Alfadhel, M. Genetic Disorders Associated with Metal Metabolism. Cells 2019, 8, 1598. [Google Scholar] [CrossRef]
- Ye, R.; Huang, J.; Wang, Z.; Chen, Y.; Dong, Y. The Role and Mechanism of Essential Selenoproteins for Homeostasis. Antioxidants 2022, 11, 973. [Google Scholar] [CrossRef]
- Lu, Z.; Marks, E.; Chen, J.; Moline, J.; Barrows, L.; Raisbeck, M.; Volitakis, I.; Cherny, R.A.; Chopra, V.; Bush, A.I.; et al. Altered selenium status in Huntington’s disease: Neuroprotection by selenite in the N171-82Q mouse model. Neurobiol. Dis. 2014, 71, 34–42. [Google Scholar] [CrossRef]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| HD Patients | Controls | p-Value | |
|---|---|---|---|
| Number of subjects | 14 | 14 | n/a |
| Age, Mean (SD) | 54.64 (12.39) | 78.5 (13.46) | <0.0001 |
| Individual age in years: | |||
| Patient/control–1 | 70 | 84 | |
| Patient/control–2 | 57 | 84 | |
| Patient/control–3 | 51 | 81 | |
| Patient/control–4 | 52 | 87 | |
| Patient/control–5 | 67 | 90 | |
| Patient/control–6 | 51 | 89 | |
| Patient/control–7 | 33 | 89 | |
| Patient/control–8 | 47 | 54 | |
| Patient/control–9 | 48 | 53 | |
| Patient/control–10 | na | 84 | |
| Patient/control–11 | 50 | 60 | |
| Patient/control–12 | 68 | 89 | |
| Patient/control–13 | 72 | 83 | |
| Patient/control–14 | 75 | 90 | |
| Sex | |||
| Males | 8 (57.1) | 8 (57.1) | 0.45 |
| Females | 6 (42.8) | 6 (42.8) | |
| Postmortem delay (PMD)-Minutes | |||
| Mean (SD) | 77.35 (71.63) | 69.28 (38.09) | 0.65 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vishweswaraiah, S.; Yilmaz, A.; Saiyed, N.; Khalid, A.; Koladiya, P.R.; Pan, X.; Macias, S.; Robinson, A.C.; Mann, D.; Green, B.D.; et al. Integrative Analysis Unveils the Correlation of Aminoacyl-tRNA Biosynthesis Metabolites with the Methylation of the SEPSECS Gene in Huntington’s Disease Brain Tissue. Genes 2023, 14, 1752. https://doi.org/10.3390/genes14091752
Vishweswaraiah S, Yilmaz A, Saiyed N, Khalid A, Koladiya PR, Pan X, Macias S, Robinson AC, Mann D, Green BD, et al. Integrative Analysis Unveils the Correlation of Aminoacyl-tRNA Biosynthesis Metabolites with the Methylation of the SEPSECS Gene in Huntington’s Disease Brain Tissue. Genes. 2023; 14(9):1752. https://doi.org/10.3390/genes14091752
Chicago/Turabian StyleVishweswaraiah, Sangeetha, Ali Yilmaz, Nazia Saiyed, Abdullah Khalid, Purvesh R. Koladiya, Xiaobei Pan, Shirin Macias, Andrew C. Robinson, David Mann, Brian D. Green, and et al. 2023. "Integrative Analysis Unveils the Correlation of Aminoacyl-tRNA Biosynthesis Metabolites with the Methylation of the SEPSECS Gene in Huntington’s Disease Brain Tissue" Genes 14, no. 9: 1752. https://doi.org/10.3390/genes14091752