
Investigation of lncRNA in Bos taurus Mammary Tissue during Dry and Lactation Periods

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Sequence Assembly and Mapping
2.3. lncRNA Identification
2.4. Comparison to Coding Transcripts
2.5. Differential Expression Analysis
2.6. Phastcon Analysis
2.7. Transcriptional Annotation Based on Sequence Homology
2.8. Gene Co-Expression Correlation and Ontology
2.9. Transcriptional Annotation Based on Correlation
2.10. Heritability Enrichment Analysis
- b: a vector of fixed effects including intercept
- X: a covariate matrix corresponding to b, , and : a vector of random effects representing the genetic contributions of the variants within lncRNAs
- : a vector of random effects representing the genetic contributions of the variants within the remaining genome
- e: a vector of residual effects
- : a genomic relationship matrix (GRM) constructed by genotypes corresponding to lncRNAs
- : a genomic relationship matrix (GRM) constructed by genotypes corresponding to the remaining genome
3. Results
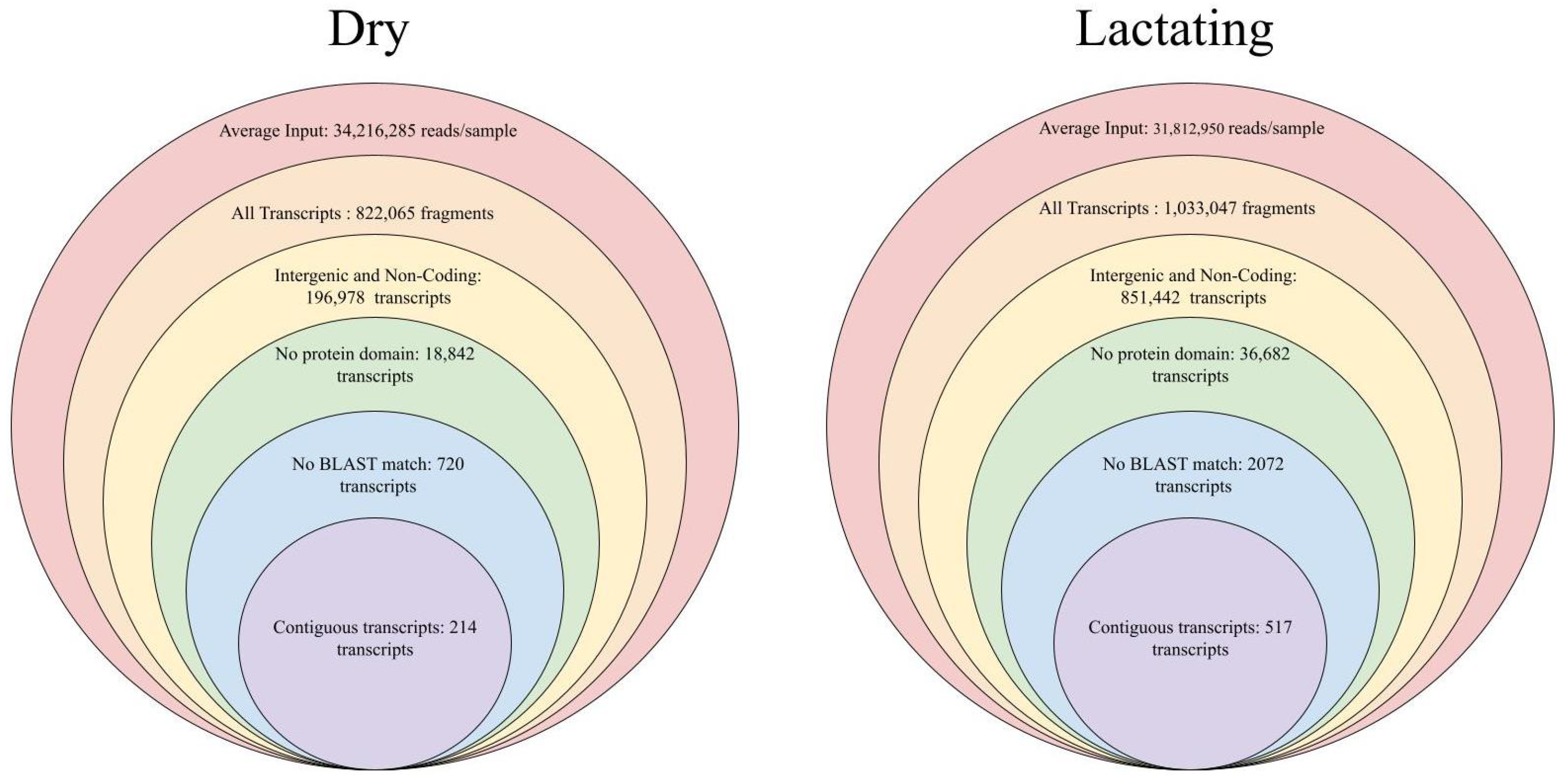
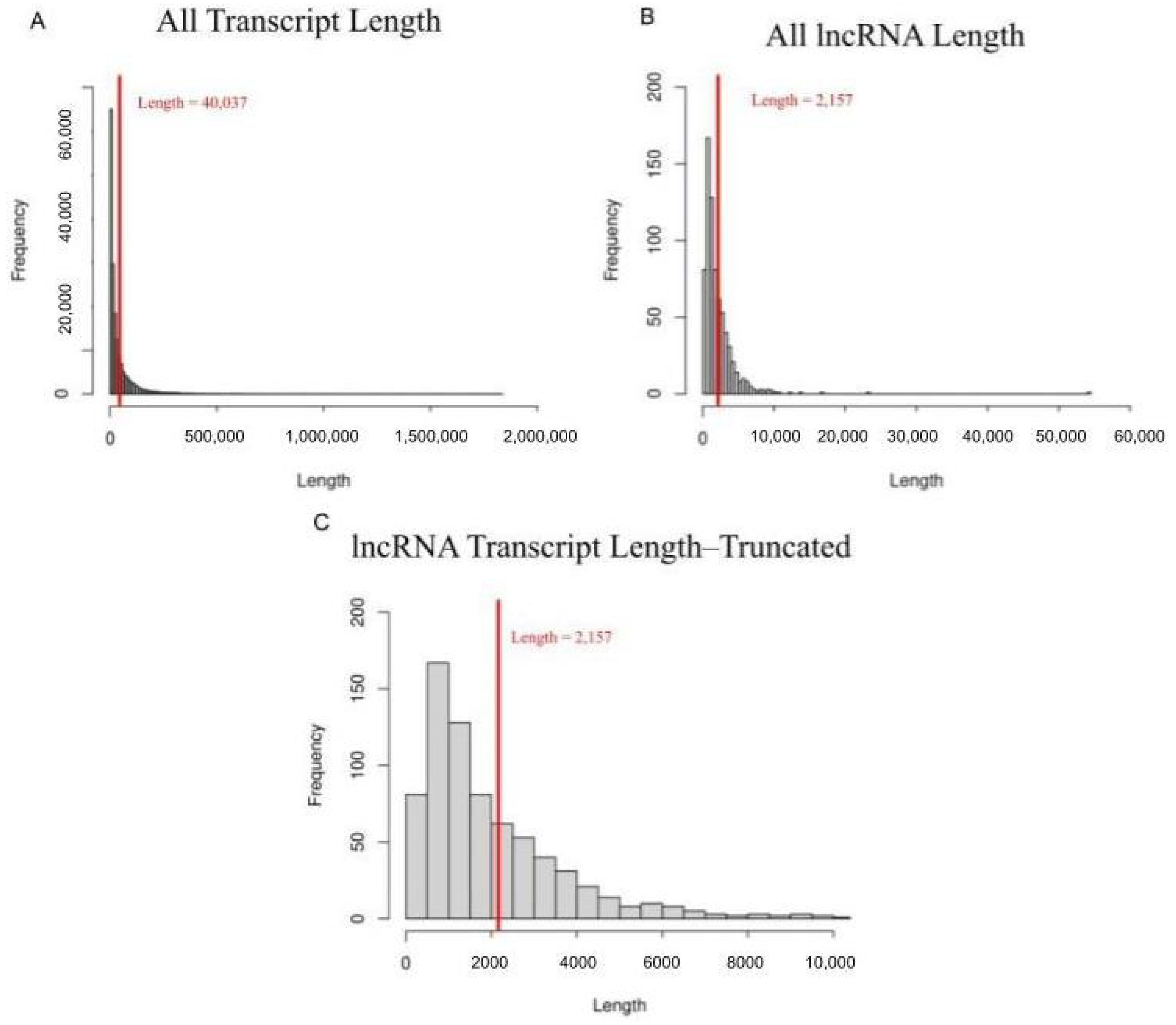
3.1. lncRNA Identification
3.2. lncRNAs as Compared to Protein Coding Transcripts
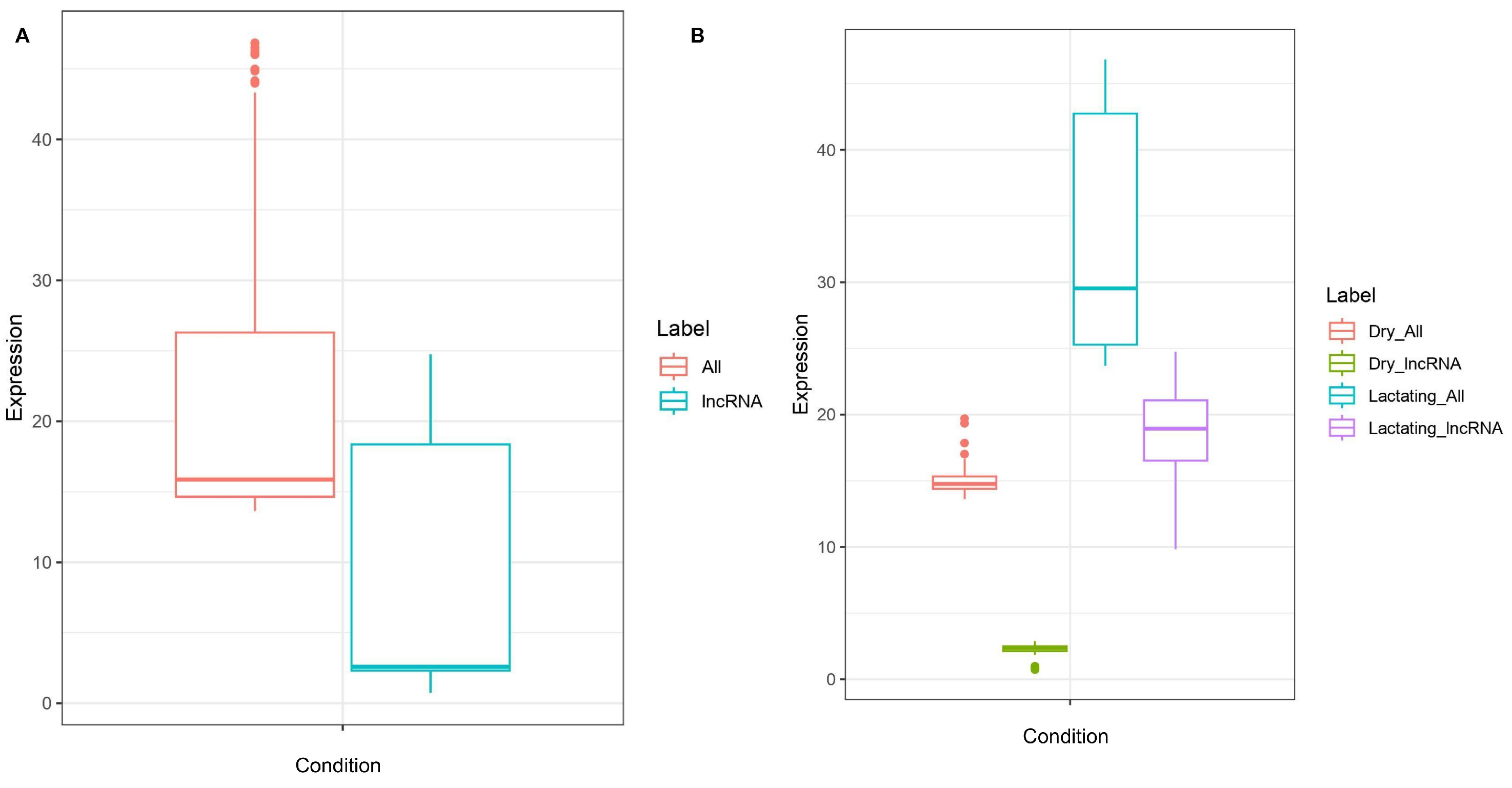
3.3. Differential Expression of lncRNA in Dry Versus Lactating Mammary Tissue
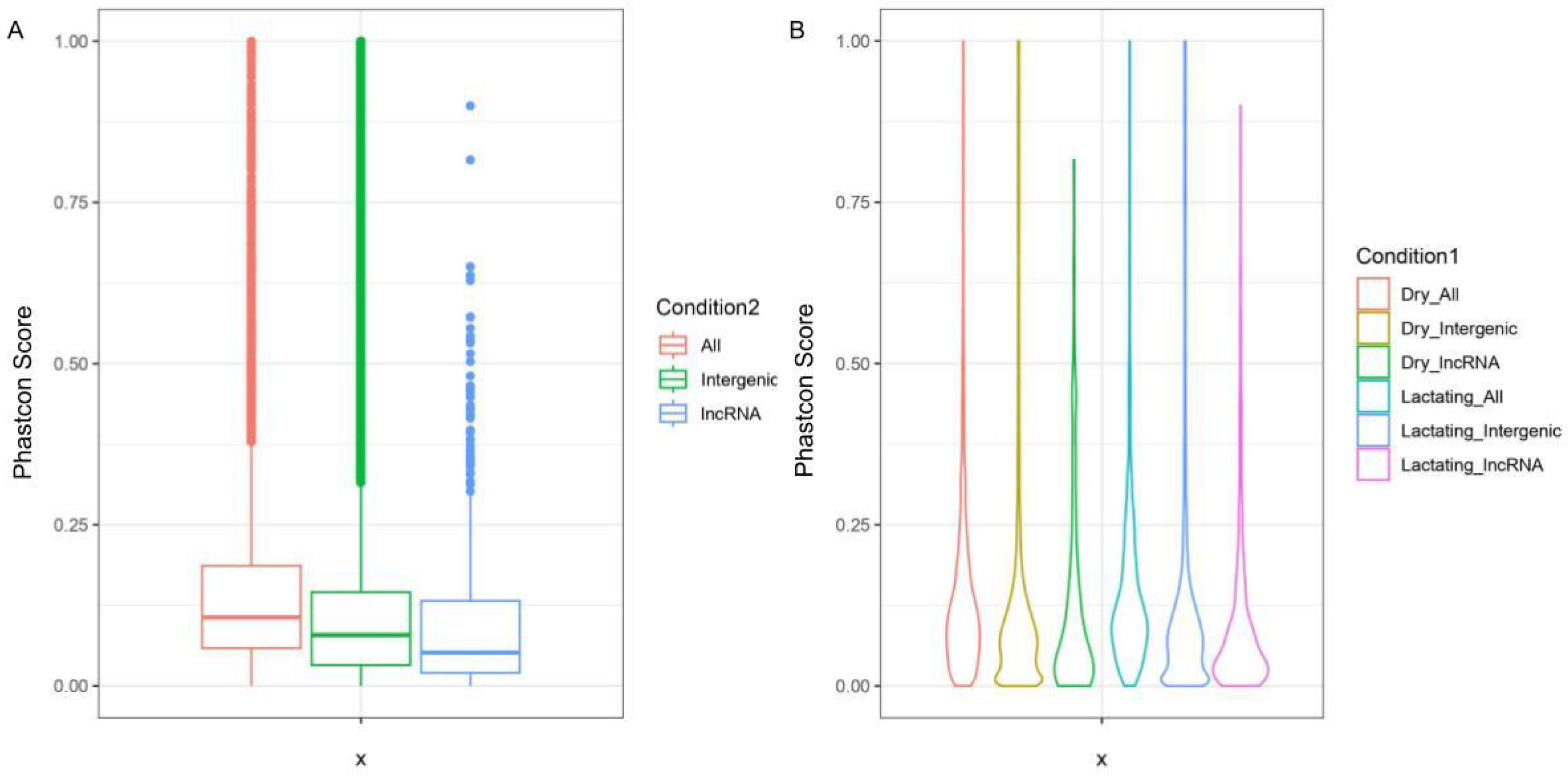
3.4. Sequence Conservation of lncRNA
3.5. Annotation of lncRNA of Interest Based on Homology
3.6. lncRNA-Gene Coexpression Correlation and Ontology
3.7. lncRNA-Gene Coexpression Correlation Annotation
3.8. SNP Heritability Enrichment Analysis on Cattle Traits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kung, J.T.; Colognori, D.; Lee, J.T. Long noncoding RNAs: Past, present, and future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Josset, L.; Tchitchek, N.; Gralinski, L.E.; Ferris, M.T.; Eisfeld, A.J.; Green, R.R.; Thomas, M.J.; Tisoncik-Go, J.; Schroth, G.P.; Kawaoka, Y.; et al. Annotation of long non-coding RNAs expressed in Collaborative Cross founder mice in response to respiratory virus infection reveals a new class of interferon-stimulated transcripts. RNA Biol. 2014, 11, 875. [Google Scholar] [CrossRef] [PubMed]
- Kern, C.; Wang, Y.; Chitwood, J.; Korf, I.; Delany, M.; Cheng, H.; Medrano, J.F.; Van Eenennaam, A.L.; Ernst, C.; Ross, P.; et al. Genome-wide identification of tissue-specific long non-coding RNA in three farm animal species 06 Biological Sciences 0604 Genetics. BMC Genom. 2018, 19, 684. [Google Scholar] [CrossRef]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. Lncbook: A curated knowledgebase of human long non-coding rnas. Nucleic Acids Res. 2019, 47, D128–D134. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Fang, L.; Baldwin, R.L.; Connor, E.E.; Cole, J.B.; Van Tassell, C.P.; Ma, L.; Li, C.J.; Liu, G.E. Single-cell transcriptomic analyses of dairy cattle ruminal epithelial cells during weaning. Genomics 2021, 113, 2045–2055. [Google Scholar] [CrossRef]
- Jia, X.; He, Y.; Chen, S.Y.; Wang, J.; Hu, S.; Lai, S.J. Genome-wide identification and characterisation of long non-coding RNAs in two Chinese cattle breeds. Ital. J. Anim. Sci. 2020, 19, 383–391. [Google Scholar] [CrossRef]
- Koufariotis, L.T.; Chen, Y.P.P.; Chamberlain, A.; Vander Jagt, C.; Hayes, B.J. A catalogue of novel bovine long noncoding RNA across 18 tissues. PLoS ONE 2015, 10, e0141225. [Google Scholar] [CrossRef]
- Wang, J.; Sun, H.Z.; O’Hara, E.; Chen, H.; Guan, L. 20 Profiling circRNA and lncRNA Expression in Key Bovine Metabolic Tissues. J. Anim. Sci. 2021, 99, 9. [Google Scholar] [CrossRef]
- Nolte, W.; Weikard, R.; Brunner, R.M.; Albrecht, E.; Hammon, H.M.; Reverter, A.; Kühn, C. Biological Network Approach for the Identification of Regulatory Long Non-Coding RNAs Associated With Metabolic Efficiency in Cattle. Front. Genet. 2019, 10, 1130. [Google Scholar] [CrossRef]
- Finot, L.; Chanat, E.; Dessauge, F. Bovine mammary gland development: New insights into the epithelial hierarchy. bioRxiv 2018, 251637. [Google Scholar] [CrossRef]
- Khan, Y.S.; Sajjad, H. Anatomy, Thorax, Mammary Gland. StatPearls Publishing. 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK547666/ (accessed on 10 January 2022).
- Nguyen, L.T.; Reverter, A.; Cánovas, A.; Venus, B.; Anderson, S.T.; Islas-Trejo, A.; Dias, M.M.; Crawford, N.F.; Lehnert, S.A.; Medrano, J.F.; et al. STAT6, PBX2, and PBRM1 emerge as predicted regulators of 452 differentially expressed genes associated with puberty in Brahman heifers. Front. Genet. 2018, 9, 87. [Google Scholar] [CrossRef]
- Dado-Senn, B.; Skibiel, A.L.; Fabris, T.F.; Zhang, Y.; Dahl, G.E.; Peñagaricano, F.; Laporta, J. RNA-Seq reveals novel genes and pathways involved in bovine mammary involution during the dry period and under environmental heat stress. Sci. Rep. 2018, 8, 11096. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.T.; Zou, Y.X.; White, R.R.; Liu, J.X.; Liu, H.Y. Transcriptomic profiles of the bovine mammary gland during lactation and the dry period. Funct. Integr. Genom. 2018, 18, 125–140. [Google Scholar] [CrossRef]
- GitHub-ncbi/sra-Tools: SRA Tools. Available online: https://github.com/ncbi/sra-tools (accessed on 1 October 2022).
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- ARS-UCD1.2-bosTau9-Genome-Assembly-NCBI. Available online: https://www.ncbi.nlm.nih.gov/assembly/GCF_002263795.1/ (accessed on 1 October 2022).
- StringTie. Available online: http://ccb.jhu.edu/software/stringtie/index.shtml?t=manual (accessed on 1 December 2022).
- Bos_taurus_UMD_3.1.1-Genome-Assembly-NCBI. Available online: https://www.ncbi.nlm.nih.gov/assembly/GCF_000003055.6/ (accessed on 1 October 2022).
- Cufflinks. Available online: http://cole-trapnell-lab.github.io/cufflinks/cuffcompare/ (accessed on 1 December 2022).
- CPC2 @ CBI, PKU. Available online: http://cpc2.gao-lab.org/download.php (accessed on 1 December 2022).
- Pfam: Search Pfam. Available online: http://pfam.xfam.org/search#tabview=tab1 (accessed on 1 January 2023).
- Nucleotide BLAST: Search Nucleotide Databases Using a Nucleotide Query. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome (accessed on 1 January 2023).
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 1 March 2023).
- Lift Genome Annotations. Available online: https://genome.ucsc.edu/cgi-bin/hgLiftOver (accessed on 1 January 2023).
- Hg38 100-Way Conservation Alignment-Genomewiki. Available online: http://genomewiki.ucsc.edu/index.php/Hg38_100-way_conservation_alignment (accessed on 1 January 2023).
- multiBigwigSummary—DeepTools 3.5.2 Documentation. Available online: https://deeptools.readthedocs.io/en/develop/content/tools/multiBigwigSummary.html (accessed on 1 March 2023).
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Overview-Salmon: Fast, Accurate and Bias-Aware Transcript Quantification from RNA-Seq Data. Available online: https://combine-lab.github.io/salmon/ (accessed on 1 April 2023).
- Gene Ontology Resource. Available online: http://geneontology.org/ (accessed on 1 April 2023).
- Jiang, J.; Cole, J.B.; Freebern, E.; Da, Y.; VanRaden, P.M.; Ma, L. Functional annotation and Bayesian fine-mapping reveals candidate genes for important agronomic traits in Holstein bulls. Commun. Biol. 2019, 2, 212. [Google Scholar] [CrossRef]
- Freebern, E.; Santos, D.J.A.; Fang, L.; Jiang, J.; Parker Gaddis, K.L.; Liu, G.E.; VanRaden, P.M.; Maltecca, C.; Cole, J.B.; Ma, L. GWAS and fine-mapping of livability and six disease traits in Holstein cattle. BMC Genom. 2020, 21, 41. [Google Scholar] [CrossRef] [PubMed]
- GitHub—jiang18/mph: MINQUE for Partitioning Heritability. Available online: https://github.com/jiang18/mph (accessed on 1 June 2023).
- National Center for Biotechnology Information (NCBI). Available online: https://www.ncbi.nlm.nih.gov/gene/ (accessed on 1 April 2023).
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- He, Y.; Ding, Y.; Zhan, F.; Zhang, H.; Han, B.; Hu, G.; Zhao, K.; Yang, N.; Yu, Y.; Mao, L.; et al. The conservation and signatures of lincRNAs in Marek’s disease of chicken. Sci. Rep. 2015, 5, 15184. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [PubMed]
- Johnsson, P.; Lipovich, L.; Grandér, D.; Morris, K.V. Evolutionary conservation of long noncoding RNAs; sequence, structure, function. Biochim. Biophys. Acta 2014, 1840, 1063. [Google Scholar] [CrossRef] [PubMed]
- Aldahmash, A.; Lunny, D.; Lane, B. Morphogenesis of the mammary gland and the role of keratin expression. Breast Cancer Res. 2005, 7, P3.09. [Google Scholar] [CrossRef]
- Lukowski, S.W.; Lloyd-Jones, L.R.; Holloway, A.; Kirsten, H.; Hemani, G.; Yang, J.; Small, K.; Zhao, J.; Metspalu, A.; Dermitzakis, E.T.; et al. Genetic correlations reveal the shared genetic architecture of transcription in human peripheral blood. Nat. Commun. 2017, 8, 483. [Google Scholar] [CrossRef]
- Cameron, E.L. Pregnancy and olfaction: A review. Front. Psychol. 2014, 5, 67. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dry | Lactating | p-Value | ||
|---|---|---|---|---|
| lncRNA | Expression | lncRNA | Expression | |
| chr1: 88,402,354–88,403,436 | 0.552 | chr1: 88,402,258–88,403,346 | 0.436 | 0.2258 |
| chr10: 28,036,993–28,037,708 | 7.006 | chr10: 28,036,875–28,037,846 | 4.436 | < |
| chr11: 102,644,124–102,648,416 | 0.592 | chr11: 102,644,428–102,648,357 | 0.374 | |
| chr11: 74,657,704–74,660,914 | 0.325 | chr11: 74,658,354–74,660,959 | 0.281 | 0.06305 |
| chr12: 32,163,152–32,163,433 | 0.953 | chr12: 32,163,199–32,163,464 | 0.966 | 0.8596 |
| chr12: 84,995,822–84,996,199 | 19.591 | chr12: 84,995,788–84,996,023 | 16.090 | 0.03071 |
| chr13: 29,201,242–29,205,645 | 0.305 | chr13: 29,202,525–29,205,728 | 0.554 | |
| chr13: 63,992,771–63,994,410 | 0.750 | chr13: 63,992,252–63,994,789 | 0.310 | |
| chr13: 76,610,578–76,612,313 | 0.617 | chr13: 76,610,635–76,611,936 | 0.562 | 0.4465 |
| chr14: 17,032–17,554 | 0.585 | chr14: 16,830–18,164 | 0.733 | 0.001775 |
| chr16: 42,802,614–42,812,241 | 33.971 | chr16: 42,802,637–42,812,194 | 15.692 | < |
| chr16: 48,586,547–48,586,896 | 0.458 | chr16: 48,584,486–48,586,868 | 0.612 | 0.0005258 |
| chr16: 71,804,283–71,805,220 | 1.440 | chr16: 71,804,288–71,805,212 | 18.671 | < |
| chr17: 69,703,586–69,705,950 | 2.227 | chr17: 69,703,579–69,705,885 | 1.975 | 0.3815 |
| chr18: 13,007,057–13,010,244 | 5.469 | chr18: 13,007,179–13,009,811 | 28.722 | |
| chr18: 1,829,126–1,830,665 | 0.196 | chr18: 1,829,460–1,830,200 | 0.398 | 0.0001832 |
| chr18: 38,105,619–38,111,753 | 6.826 | chr18: 38,105,601–38,111,755 | 3.175 | < |
| chr18: 58,480,168–58,480,492 | 0.326 | chr18: 58,479,948–58,480,580 | 0.448 | 0.0004047 |
| chr18: 7,018,326–7,020,326 | 0.521 | chr18: 7,018,199–7,020,326 | 0.283 | |
| chr19: 41,517,108–41,519,655 | 0.550 | chr19: 41,518,998–41,519,227 | 0.288 | |
| chr2: 111,874,235–111,875,148 | 4.087 | chr2: 111,874,066–111,877,333 | 2.759 | |
| chr2: 115,539,609–115,540,162 | 235.756 | chr2: 115,539,562–115,540,164 | 138.488 | |
| chr2: 125,783,114–125,794,043 | 24.285 | chr2: 125,777,445–125,793,948 | 33.604 | |
| chr21: 58,272,796–58,274,409 | 1.584 | chr21: 58,272,808–58,274,430 | 0.854 | < |
| chr21: 68,475,571–68,478,473 | 2.627 | chr21: 68,475,747–68,477,894 | 2.432 | 0.6144 |
| chr23: 11,415,564–11,420,435 | 19.060 | chr23: 11,415,556–11,420,435 | 8.688 | < |
| chr23: 17,153,445–17,154,227 | 8.728 | chr23: 17,153,446–17,154,226 | 10.971 | 0.0002695 |
| chr23: 17,771,969–17,776,464 | 2.292 | chr23: 17,771,923–17,776,476 | 0.578 | < |
| chr25: 1,035,522–1,036,687 | 10.186 | chr25: 1,035,522–1,036,586 | 16.318 | |
| chr25: 35,533,291–35,534,130 | 0.988 | chr25: 35,533,317–35,534,096 | 19.412 | < |
| chr25: 39,512,349–39,513,122 | 12.600 | chr25: 39,512,418–39,513,043 | 5.562 | < |
| chr25: 488,853–489,320 | 3.302 | chr25: 488,852–489,379 | 2.741 | 0.02195 |
| chr25: 786,947–787,788 | 1.176 | chr25: 786,326–788,530 | 0.780 | |
| chr26: 10,508,859–10,510,408 | 0.425 | chr26: 10,508,507–10,510,415 | 0.526 | |
| chr26: 21,485,237–21,487,166 | 0.418 | chr26: 21,485,168–21,487,115 | 1.136 | |
| chr26: 51,014,183–51,020,151 | 0.424 | chr26: 51,016,793–51,020,761 | 0.234 | |
| chr27: 1,092,242–1,093,287 | 0.894 | chr27: 1,092,247–1,093,275 | 1.036 | 0.08457 |
| chr29: 46,399,007–46,401,176 | 0.316 | chr29: 46,399,026–46,401,485 | 0.302 | 0.745 |
| chr29: 46,623,288–46,624,946 | 1.844 | chr29: 46,623,270–46,625,478 | 3.484 | |
| chr29: 47,279,192–47,281,419 | 0.440 | chr29: 47,278,057–47,281,321 | 1.610 | < |
| chr29: 47,437,521–47,440,208 | 0.603 | chr29: 47,437,552–47,440,146 | 1.012 | |
| chr4: 52,093,644–52,094,267 | 4.437 | chr4: 52,093,643–52,094,277 | 0.438 | |
| chr5: 118,095,776–118,100,684 | 0.434 | chr5: 118,095,874–118,100,834 | 0.303 | |
| chr6: 3,400,389–3,401,869 | 0.596 | chr6: 3,400,394–3,401,718 | 5.301 | < |
| chr6: 62,052,709–62,053,912 | 0.368 | chr6: 62,052,696–62,054,059 | 0.511 | 0.002736 |
| chr7: 10,013,140–10,014,516 | 1.509 | chr7: 10,013,140–10,014,265 | 0.391 | |
| chr7: 11,559,071–11,560,742 | 0.058 | chr7: 11,559,861–11,561,374 | 0.100 | 0.2047 |
| chr7: 17,181,130–17,183,216 | 0.703 | chr7: 17,181,196–17,183,208 | 11.387 | < |
| chr7: 60,229,378–60,232,948 | 9.270 | chr7: 60,229,471–60,232,796 | 4.910 | |
| chr9: 98,186,197–98,187,338 | 1.152 | chr9: 98,186,218–98,187,339 | 3.450 | < |
| ChrUn.004.1159: 35,363–37,180 | 0.171 | ChrUn.004.1159: 35,361–38,038 | 0.344 | |
| ChrUn.004.301: 120,666–127,066 | 0.410 | ChrUn.004.301: 124,684–128,068 | 0.170 | |
| ChrUn.004.3283: 1785–2847 | 0.330 | ChrUn.004.3283: 1822–2792 | 2.147 |
| DPR | Livability | Milk | Stature | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Enr | SE | p | Enr | SE | p | Enr | SE | p | Enr | SE | p | |
| Dry | 8.92 | 3.65 | 0.02 | 6.10 | 5.12 | 0.16 | 7.39 | 2.50 | 0.01 | 2.93 | 2.27 | 0.20 |
| Lact | 4.96 | 1.91 | 0.02 | −0.39 | 2.39 | 0.72 | 4.73 | 1.36 | 6.10 | 1.53 | 0.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marceau, A.; Wang, J.; Iqbal, V.; Jiang, J.; Liu, G.E.; Ma, L. Investigation of lncRNA in Bos taurus Mammary Tissue during Dry and Lactation Periods. Genes 2023, 14, 1789. https://doi.org/10.3390/genes14091789
Marceau A, Wang J, Iqbal V, Jiang J, Liu GE, Ma L. Investigation of lncRNA in Bos taurus Mammary Tissue during Dry and Lactation Periods. Genes. 2023; 14(9):1789. https://doi.org/10.3390/genes14091789
Chicago/Turabian StyleMarceau, Alexis, Junjian Wang, Victoria Iqbal, Jicai Jiang, George E. Liu, and Li Ma. 2023. "Investigation of lncRNA in Bos taurus Mammary Tissue during Dry and Lactation Periods" Genes 14, no. 9: 1789. https://doi.org/10.3390/genes14091789