

Mechanisms of Rapid Karyotype Evolution in Mammals

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tachytely: A Retrospective Application to Rapid Karyotype Evolution
3. Old and New Techniques in Studying Chromosome Evolution
4. Mechanisms of Rapid Karyotype Evolution
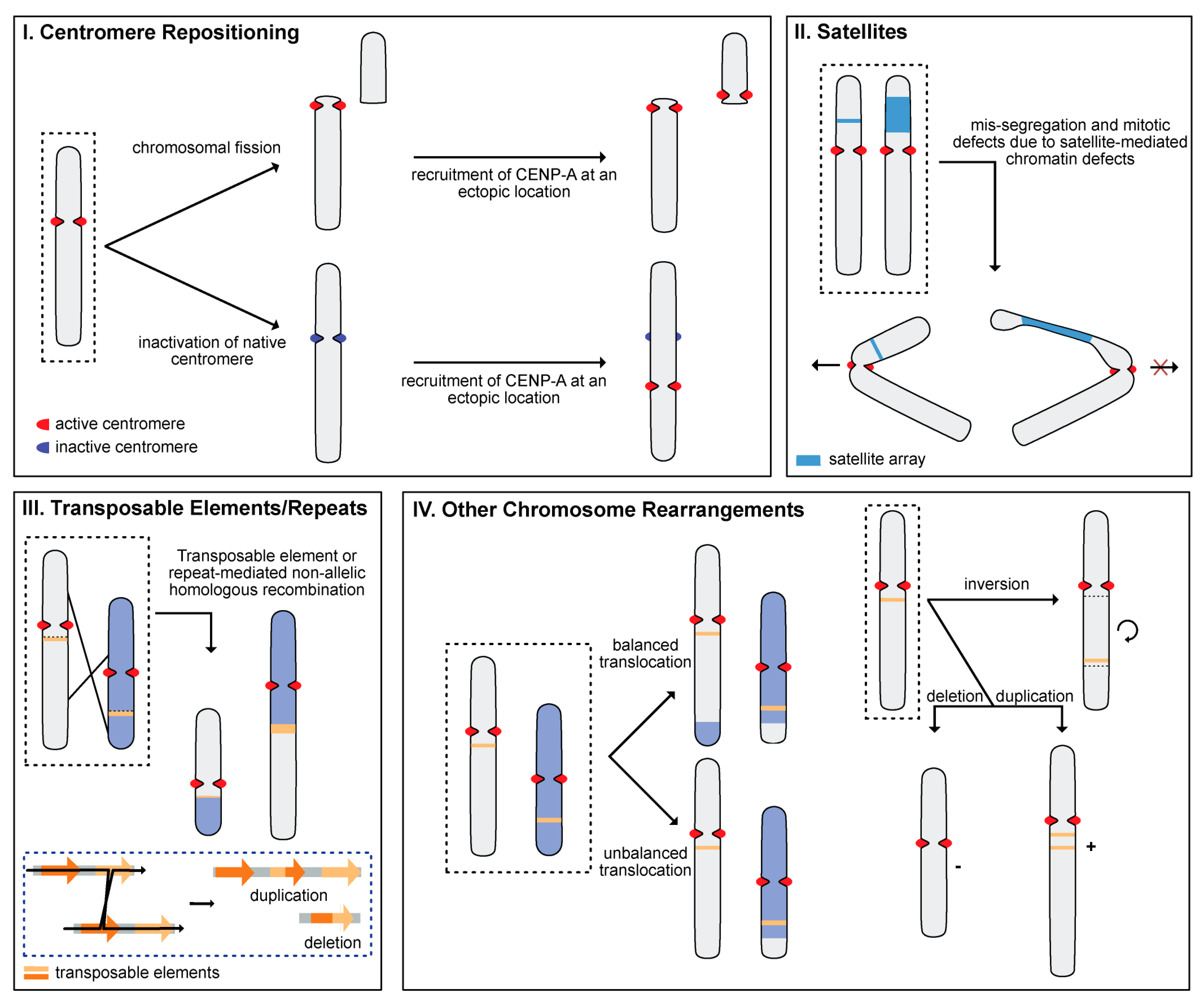
4.1. Centromere Repositioning
4.2. Satellite DNA (satDNA)
4.3. Transposable Elements (TEs)
4.4. Other Chromosome Rearrangements (CRs)
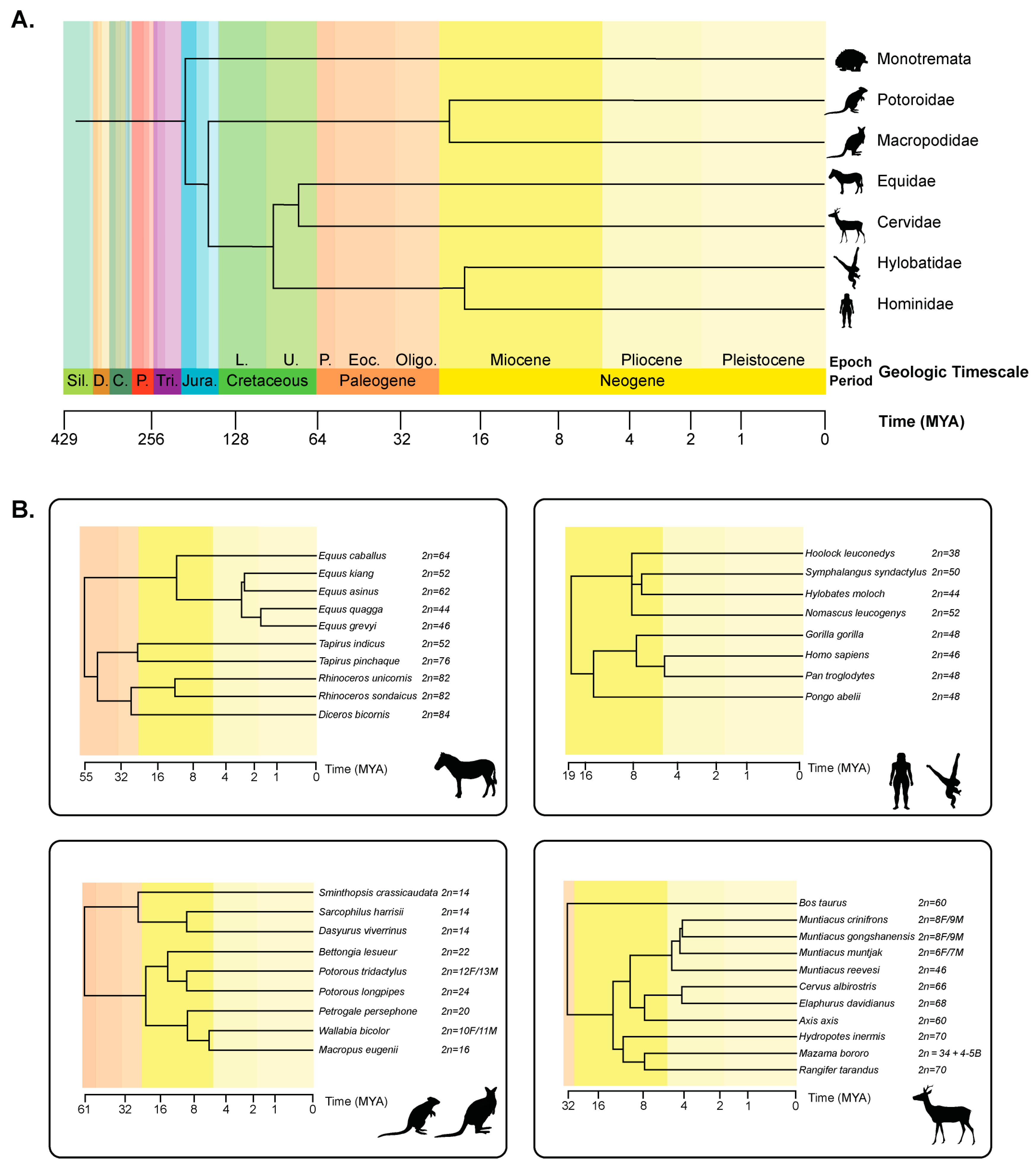
5. Lineages Characterized by Rapid Chromosome Evolution
5.1. Equidae
5.2. Hylobatidae
5.3. Macropodidae
5.4. Potoroidae
5.5. Artiodactyla: Cervidae
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fritz, A.J.; Sehgal, N.; Pliss, A.; Xu, J.; Berezney, R. Chromosome Territories and the Global Regulation of the Genome. Genes Chromosomes Cancer 2019, 58, 407–426. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhao, Y.; Shiraigol, W.; Li, B.; Bai, D.; Ye, W.; Daidiikhuu, D.; Yang, L.; Jin, B.; Zhao, Q.; et al. Analysis of Horse Genomes Provides Insight into the Diversification and Adaptive Evolution of Karyotype. Sci. Rep. 2014, 4, 4958. [Google Scholar] [CrossRef] [PubMed]
- Hartley, G.A.; Okhovat, M.; O’Neill, R.J.; Carbone, L. Comparative Analyses of Gibbon Centromeres Reveal Dynamic Genus-Specific Shifts in Repeat Composition. Mol. Biol. Evol. 2021, 38, 3972–3992. [Google Scholar] [CrossRef] [PubMed]
- Westerman, M.; Meredith, R.W.; Springer, M.S. Cytogenetics Meets Phylogenetics: A Review of Karyotype Evolution in Diprotodontian Marsupials. J. Hered. 2010, 101, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Rens, W.; O’Brien, P.C.; Yang, F.; Graves, J.A.; Ferguson-Smith, M.A. Karyotype Relationships between Four Distantly Related Marsupials Revealed by Reciprocal Chromosome Painting. Chromosome Res. 1999, 7, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Torgasheva, A.A.; Basheva, E.A.; Gómez Fernández, M.J.; Mirol, P.; Borodin, P.M. Chromosomes and Speciation in Tuco-Tuco (Ctenomys, Hystricognathi, Rodentia). Russ. J. Genet. Appl. Res. 2017, 7, 350–357. [Google Scholar] [CrossRef]
- Baker, R.J.; Bickham, J.W. Karyotypic Evolution in Bats: Evidence of Extensive and Conservative Chromosomal Evolution in Closely Related Taxa. Syst. Biol. 1980, 29, 239–253. [Google Scholar] [CrossRef]
- Sotero-Caio, C.G.; Volleth, M.; Hoffmann, F.G.; Scott, L.; Wichman, H.A.; Yang, F.; Baker, R.J. Integration of Molecular Cytogenetics, Dated Molecular Phylogeny, and Model-Based Predictions to Understand the Extreme Chromosome Reorganization in the Neotropical Genus Tonatia (Chiroptera: Phyllostomidae). BMC Evol. Biol. 2015, 15, 220. [Google Scholar] [CrossRef]
- Yin, Y.; Fan, H.; Zhou, B.; Hu, Y.; Fan, G.; Wang, J.; Zhou, F.; Nie, W.; Zhang, C.; Liu, L.; et al. Molecular Mechanisms and Topological Consequences of Drastic Chromosomal Rearrangements of Muntjac Deer. Nat. Commun. 2021, 12, 6858. [Google Scholar] [CrossRef]
- Rocchi, M.; Archidiacono, N.; Schempp, W.; Capozzi, O.; Stanyon, R. Centromere Repositioning in Mammals. Heredity 2012, 108, 59–67. [Google Scholar] [CrossRef]
- Thakur, J.; Packiaraj, J.; Henikoff, S. Sequence, Chromatin and Evolution of Satellite DNA. Int. J. Mol. Sci. 2021, 22, 4309. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G. Transposable Elements in Gene Regulation and in the Evolution of Vertebrate Genomes. Curr. Opin. Genet. Dev. 2009, 19, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G.G. Tempo and Mode in Evolution; Columbia University Press: New York, NY, USA, 1944; ISBN 9780231891721. [Google Scholar]
- Simpson, G.G. The Major Features of Evolution; Columbia University Press: New York, NY, USA, 1953; ISBN 9780231895330. [Google Scholar]
- Avise, J.C.; Nelson, W.S.; Sugita, H. A Speciational history of “Living fossils”: Molecular evolutionary patterns in horseshoe crabs. Evolution 1994, 48, 1986–2001. [Google Scholar] [CrossRef] [PubMed]
- Merino, S.; Vásquez, R.A.; Martínez, J.; Celis-Diez, J.L.; Gutiérrez-Jiménez, L.; Ippi, S.; Sánchez-Monsalvez, I.; Martínez-de La Puente, J. Molecular Characterization of an Ancient Hepatozoon Species Parasitizing the “living Fossil” Marsupial “Monito Del Monte” Dromiciops Gliroides from Chile. Biol. J. Linn. Soc. Lond. 2009, 98, 568–576. [Google Scholar] [CrossRef]
- Casane, D.; Laurenti, P. Why Coelacanths Are Not “Living Fossils”: A Review of Molecular and Morphological Data. Bioessays 2013, 35, 332–338. [Google Scholar] [CrossRef]
- Mathers, T.C.; Hammond, R.L.; Jenner, R.A.; Hänfling, B.; Gómez, A. Multiple Global Radiations in Tadpole Shrimps Challenge the Concept of “Living Fossils”. PeerJ 2013, 1, e62. [Google Scholar] [CrossRef]
- Fitch, W.M.; Ayala, F.J. Tempo and Mode in the Macroevolutionary Reconstruction of Darwinism; National Academies Press (US): Washington, DC, USA, 1995. [Google Scholar]
- Howell, W.M. Visualization of Ribosomal Gene Activity: Silver Stains Proteins Associated with rRNA Transcribed from Oocyte Chromosomes. Chromosoma 1977, 62, 361–367. [Google Scholar] [CrossRef]
- Bloom, S.E.; Goodpasture, C. An Improved Technique for Selective Silver Staining of Nucleolar Organizer Regions in Human Chromosomes. Hum. Genet. 1976, 34, 199–206. [Google Scholar] [CrossRef]
- Pardue, M.L.; Gall, J.G. Chromosomal Localization of Mouse Satellite DNA. Science 1970, 168, 1356–1358. [Google Scholar] [CrossRef]
- Wilzbach, K.E. Tritium-labeling by exposure of organic compounds to tritium gas1. J. Am. Chem. Soc. 1957, 79, 1013. [Google Scholar] [CrossRef]
- Bauman, J.G.J.; Wiegant, J.; Borst, P.; van Duijn, P. A New Method for Fluorescence Microscopical Localization of Specific DNA Sequences by in Situ Hybridization of Fluorochrome-Labelled RNA. Exp. Cell Res. 1980, 128, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Lichter, P.; Borden, J.; Ward, D.C.; Manuelidis, L. Detection of Chromosome Aberrations in Metaphase and Interphase Tumor Cells by in Situ Hybridization Using Chromosome-Specific Library Probes. Hum. Genet. 1988, 80, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Lichter, P.; Cremer, T.; Borden, J.; Manuelidis, L.; Ward, D.C. Delineation of Individual Human Chromosomes in Metaphase and Interphase Cells by in Situ Suppression Hybridization Using Recombinant DNA Libraries. Hum. Genet. 1988, 80, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Pinkel, D.; Landegent, J.; Collins, C.; Fuscoe, J.; Segraves, R.; Lucas, J.; Gray, J. Fluorescence in Situ Hybridization with Human Chromosome-Specific Libraries: Detection of Trisomy 21 and Translocations of Chromosome 4. Proc. Natl. Acad. Sci. USA 1988, 85, 9138–9142. [Google Scholar] [CrossRef]
- Sanger, F.; Air, G.M.; Barrell, B.G.; Brown, N.L.; Coulson, A.R.; Fiddes, C.A.; Hutchison, C.A.; Slocombe, P.M.; Smith, M. Nucleotide Sequence of Bacteriophage Phi X174 DNA. Nature 1977, 265, 687–695. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Talbert, P.B.; Henikoff, S. What Makes a Centromere? Exp. Cell Res. 2020, 389, 111895. [Google Scholar] [CrossRef]
- Sankoff, D. The Where and Wherefore of Evolutionary Breakpoints. J. Biol. 2009, 8, 66. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, A.Y.; Barkley, C.A.; Zhang, Y.; Zhao, X.; Gao, M.; Edmonds, M.D.; Chong, Z. Deciphering the Exact Breakpoints of Structural Variations Using Long Sequencing Reads with DeBreak. Nat. Commun. 2023, 14, 283. [Google Scholar] [CrossRef]
- Lucas, M.C.; Novoa, E.M. Long-Read Sequencing in the Era of Epigenomics and Epitranscriptomics. Nat. Methods 2023, 20, 25–29. [Google Scholar] [CrossRef]
- Bajpai, P.K.; Harel, A.; Shafir, S.; Barazani, O. Whole Genome Sequencing Reveals Footprints of Adaptive Genetic Variation in Populations of Eruca sativa. Front. Ecol. Evol. 2022, 10, 938981. [Google Scholar] [CrossRef]
- Bock, D.G.; Liu, J.; Novikova, P.; Rieseberg, L.H. Long-Read Sequencing in Ecology and Evolution: Understanding How Complex Genetic and Epigenetic Variants Shape Biodiversity. Mol. Ecol. 2023, 32, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.H.; Bender, M.A. Cytogenetics and Evolution of Primates. Ann. N. Y. Acad. Sci. 1962, 102, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Hayman, D.L.; Martin, P.G. Cytogenetics of Marsupials. In Comparative Mammalian Cytogenetics; Springer: Berlin/Heidelberg, Germany, 1969; pp. 191–217. [Google Scholar]
- Stanyon, R.; Rocchi, M.; Capozzi, O.; Roberto, R.; Misceo, D.; Ventura, M.; Cardone, M.F.; Bigoni, F.; Archidiacono, N. Primate Chromosome Evolution: Ancestral Karyotypes, Marker Order and Neocentromeres. Chromosome Res. 2008, 16, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Deakin, J.E.; O’Neill, R.J. Evolution of Marsupial Genomes. Annu. Rev. Anim. Biosci. 2020, 8, 25–45. [Google Scholar] [CrossRef] [PubMed]
- Damas, J.; Corbo, M.; Kim, J.; Turner-Maier, J.; Farré, M.; Larkin, D.M.; Ryder, O.A.; Steiner, C.; Houck, M.L.; Hall, S.; et al. Evolution of the Ancestral Mammalian Karyotype and Syntenic Regions. Proc. Natl. Acad. Sci. USA 2022, 119, e2209139119. [Google Scholar] [CrossRef]
- O’Brien, S.J.; Graphodatsky, A.S.; Perelman, P.L. Atlas of Mammalian Chromosomes; John Wiley & Sons: Hoboken, NJ, USA, 2020; ISBN 9781119418030. [Google Scholar]
- Yoda, K.; Ando, S.; Morishita, S.; Houmura, K.; Hashimoto, K.; Takeyasu, K.; Okazaki, T. Human Centromere Protein A (CENP-A) Can Replace Histone H3 in Nucleosome Reconstitution in Vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 7266–7271. [Google Scholar] [CrossRef]
- Van Hooser, A.A.; Ouspenski, I.I.; Gregson, H.C.; Starr, D.A.; Yen, T.J.; Goldberg, M.L.; Yokomori, K.; Earnshaw, W.C.; Sullivan, K.F.; Brinkley, B.R. Specification of Kinetochore-Forming Chromatin by the Histone H3 Variant CENP-A. J. Cell Sci. 2001, 114, 3529–3542. [Google Scholar] [CrossRef]
- Talbert, P.B.; Masuelli, R.; Tyagi, A.P.; Comai, L.; Henikoff, S. Centromeric Localization and Adaptive Evolution of an Arabidopsis Histone H3 Variant. Plant Cell 2002, 14, 1053–1066. [Google Scholar] [CrossRef]
- Hasson, D.; Panchenko, T.; Salimian, K.J.; Salman, M.U.; Sekulic, N.; Alonso, A.; Warburton, P.E.; Black, B.E. The Octamer Is the Major Form of CENP-A Nucleosomes at Human Centromeres. Nat. Struct. Mol. Biol. 2013, 20, 687–695. [Google Scholar] [CrossRef]
- Murillo-Pineda, M.; Valente, L.P.; Dumont, M.; Mata, J.F.; Fachinetti, D.; Jansen, L.E.T. Induction of Spontaneous Human Neocentromere Formation and Long-Term Maturation. J. Cell Biol. 2021, 220, e202007210. [Google Scholar] [CrossRef] [PubMed]
- Burrack, L.S.; Berman, J. Neocentromeres and Epigenetically Inherited Features of Centromeres. Chromosome Res. 2012, 20, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Willard, H.F. Chromosome-Specific Organization of Human Alpha Satellite DNA. Am. J. Hum. Genet. 1985, 37, 524–532. [Google Scholar] [PubMed]
- Wang, G.; Zhang, X.; Jin, W. An Overview of Plant Centromeres. J. Genet. Genom. 2009, 36, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Birchler, J.A.; Gao, Z.; Han, F. A Tale of Two Centromeres—Diversity of Structure but Conservation of Function in Plants and Animals. Funct. Integr. Genom. 2009, 9, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, M.; Cummings, R.; Yamashita, Y.M. A Conserved Function for Pericentromeric Satellite DNA. Elife 2018, 7, e34122. [Google Scholar] [CrossRef] [PubMed]
- Melters, D.P.; Bradnam, K.R.; Young, H.A.; Telis, N.; May, M.R.; Ruby, J.G.; Sebra, R.; Peluso, P.; Eid, J.; Rank, D.; et al. Comparative Analysis of Tandem Repeats from Hundreds of Species Reveals Unique Insights into Centromere Evolution. Genome Biol. 2013, 14, R10. [Google Scholar] [CrossRef]
- Voullaire, L.E.; Slater, H.R.; Petrovic, V.; Choo, K.H. A Functional Marker Centromere with No Detectable Alpha-Satellite, Satellite III, or CENP-B Protein: Activation of a Latent Centromere? Am. J. Hum. Genet. 1993, 52, 1153–1163. [Google Scholar]
- Amor, D.J.; Bentley, K.; Ryan, J.; Perry, J.; Wong, L.; Slater, H.; Choo, K.H.A. Human Centromere Repositioning “in Progress”. Proc. Natl. Acad. Sci. USA 2004, 101, 6542–6547. [Google Scholar] [CrossRef]
- Tolomeo, D.; Capozzi, O.; Stanyon, R.R.; Archidiacono, N.; D’Addabbo, P.; Catacchio, C.R.; Purgato, S.; Perini, G.; Schempp, W.; Huddleston, J.; et al. Epigenetic origin of evolutionary novel centromeres. Sci. Rep. 2017, 7, 41980. [Google Scholar] [CrossRef]
- Earnshaw, W.C.; Rothfield, N. Identification of a Family of Human Centromere Proteins Using Autoimmune Sera from Patients with Scleroderma. Chromosoma 1985, 91, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Valdivia, M.M.; Brinkley, B.R. Fractionation and Initial Characterization of the Kinetochore from Mammalian Metaphase Chromosomes. J. Cell Biol. 1985, 101, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Naughton, C.; Huidobro, C.; Catacchio, C.R.; Buckle, A.; Grimes, G.R.; Nozawa, R.-S.; Purgato, S.; Rocchi, M.; Gilbert, N. Human Centromere Repositioning Activates Transcription and Opens Chromatin Fibre Structure. Nat. Commun. 2022, 13, 5609. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; He, X. Centromere Repositioning Causes Inversion of Meiosis and Generates a Reproductive Barrier. Proc. Natl. Acad. Sci. USA 2019, 116, 21580–21591. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.; Archidiacono, N.; Rocchi, M. Centromere Emergence in Evolution. Genome Res. 2001, 11, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Ventura, M.; Weigl, S.; Carbone, L.; Cardone, M.F.; Misceo, D.; Teti, M.; D’Addabbo, P.; Wandall, A.; Björck, E.; de Jong, P.J.; et al. Recurrent Sites for New Centromere Seeding. Genome Res. 2004, 14, 1696–1703. [Google Scholar] [CrossRef] [PubMed]
- Eder, V.; Ventura, M.; Ianigro, M.; Teti, M.; Rocchi, M.; Archidiacono, N. Chromosome 6 Phylogeny in Primates and Centromere Repositioning. Mol. Biol. Evol. 2003, 20, 1506–1512. [Google Scholar] [CrossRef]
- Cardone, M.F.; Lomiento, M.; Teti, M.G.; Misceo, D.; Roberto, R.; Capozzi, O.; D’Addabbo, P.; Ventura, M.; Rocchi, M.; Archidiacono, N. Evolutionary History of Chromosome 11 Featuring Four Distinct Centromere Repositioning Events in Catarrhini. Genomics 2007, 90, 35–43. [Google Scholar] [CrossRef]
- Ventura, M.; Antonacci, F.; Cardone, M.F.; Stanyon, R.; D’Addabbo, P.; Cellamare, A.; Sprague, L.J.; Eichler, E.E.; Archidiacono, N.; Rocchi, M. Evolutionary Formation of New Centromeres in Macaque. Science 2007, 316, 243–246. [Google Scholar] [CrossRef]
- Ferreri, G.C.; Liscinsky, D.M.; Mack, J.A.; Eldridge, M.D.B.; O’Neill, R.J. Retention of Latent Centromeres in the Mammalian Genome. J. Hered. 2005, 96, 217–224. [Google Scholar] [CrossRef]
- Carbone, L.; Nergadze, S.G.; Magnani, E.; Misceo, D.; Francesca Cardone, M.; Roberto, R.; Bertoni, L.; Attolini, C.; Francesca Piras, M.; de Jong, P.; et al. Evolutionary Movement of Centromeres in Horse, Donkey, and Zebra. Genomics 2006, 87, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Ramos, M.A. Satellite DNA: An Evolving Topic. Genes 2017, 8, 230. [Google Scholar] [CrossRef] [PubMed]
- Pös, O.; Radvanszky, J.; Buglyó, G.; Pös, Z.; Rusnakova, D.; Nagy, B.; Szemes, T. DNA Copy Number Variation: Main Characteristics, Evolutionary Significance, and Pathological Aspects. Biomed. J. 2021, 44, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Ferree, P.M.; Barbash, D.A. Species-Specific Heterochromatin Prevents Mitotic Chromosome Segregation to Cause Hybrid Lethality in Drosophila. PLoS Biol. 2009, 7, e1000234. [Google Scholar] [CrossRef] [PubMed]
- Mestrović, N.; Castagnone-Sereno, P.; Plohl, M. Interplay of Selective Pressure and Stochastic Events Directs Evolution of the MEL172 Satellite DNA Library in Root-Knot Nematodes. Mol. Biol. Evol. 2006, 23, 2316–2325. [Google Scholar] [CrossRef]
- Mravinac, B.; Plohl, M. Parallelism in Evolution of Highly Repetitive DNAs in Sibling Species. Mol. Biol. Evol. 2010, 27, 1857–1867. [Google Scholar] [CrossRef]
- Pons, J.; Gillespie, R.G. Common Origin of the Satellite DNAs of the Hawaiian Spiders of the Genus Tetragnatha: Evolutionary Constraints on the Length and Nucleotide Composition of the Repeats. Gene 2003, 313, 169–177. [Google Scholar] [CrossRef]
- Lorite, P.; Carrillo, J.A.; Tinaut, A.; Palomeque, T. Evolutionary Dynamics of Satellite DNA in Species of the Genus Formica (Hymenoptera, Formicidae). Gene 2004, 332, 159–168. [Google Scholar] [CrossRef]
- Harrison, G.E.; Heslop-Harrison, J.S. Centromeric Repetitive DNA Sequences in the Genus Brassica. Theor. Appl. Genet. 1995, 90, 157–165. [Google Scholar] [CrossRef]
- Koga, A.; Hashimoto, K.; Honda, Y.; Nishihara, H. Marsupial Genome Analysis Suggests That Satellite DNA Formation from Walb Endogenous Retrovirus Is an Event Specific to the Red-Necked Wallaby. Genes Cells 2023, 28, 149–155. [Google Scholar] [CrossRef]
- Hayashi, S.; Honda, Y.; Kanesaki, E.; Koga, A. Marsupial Satellite DNA as Faithful Reflections of Long-Terminal Repeat Retroelement Structure. Genome 2022, 65, 469–478. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. The Origin and Behavior of Mutable Loci in Maize. Proc. Natl. Acad. Sci. USA 1950, 36, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.J.; O’Neill, R.J. Transposable Elements: Genome Innovation, Chromosome Diversity, and Centromere Conflict. Chromosome Res. 2018, 26, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Marcon, H.S.; Domingues, D.S.; Silva, J.C.; Borges, R.J.; Matioli, F.F.; Fontes, M.R.d.M.; Marino, C.L. Transcriptionally Active LTR Retrotransposons in Eucalyptus Genus Are Differentially Expressed and Insertionally Polymorphic. BMC Plant Biol. 2015, 15, 198. [Google Scholar] [CrossRef] [PubMed]
- Sanseverino, W.; Hénaff, E.; Vives, C.; Pinosio, S.; Burgos-Paz, W.; Morgante, M.; Ramos-Onsins, S.E.; Garcia-Mas, J.; Casacuberta, J.M. Transposon Insertions, Structural Variations, and SNPs Contribute to the Evolution of the Melon Genome. Mol. Biol. Evol. 2015, 32, 2760–2774. [Google Scholar] [CrossRef] [PubMed]
- Landry, J.R.; Medstrand, P.; Mager, D.L. Repetitive Elements in the 5’ Untranslated Region of a Human Zinc-Finger Gene Modulate Transcription and Translation Efficiency. Genomics 2001, 76, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.K.; Han, K.; Wang, J.; Lee, J.; Wang, H.; Callinan, P.A.; Dyer, M.; Cordaux, R.; Liang, P.; Batzer, M.A. Human Genomic Deletions Mediated by Recombination between Alu Elements. Am. J. Hum. Genet. 2006, 79, 41–53. [Google Scholar] [CrossRef]
- Platt, R.N., 2nd; Vandewege, M.W.; Ray, D.A. Mammalian Transposable Elements and Their Impacts on Genome Evolution. Chromosome Res. 2018, 26, 25–43. [Google Scholar] [CrossRef]
- Casacuberta, E.; González, J. The Impact of Transposable Elements in Environmental Adaptation. Mol. Ecol. 2013, 22, 1503–1517. [Google Scholar] [CrossRef]
- Schrader, L.; Schmitz, J. The Impact of Transposable Elements in Adaptive Evolution. Mol. Ecol. 2019, 28, 1537–1549. [Google Scholar] [CrossRef]
- Suh, A.; Churakov, G.; Ramakodi, M.P.; Platt, R.N., 2nd; Jurka, J.; Kojima, K.K.; Caballero, J.; Smit, A.F.; Vliet, K.A.; Hoffmann, F.G.; et al. Multiple Lineages of Ancient CR1 Retroposons Shaped the Early Genome Evolution of Amniotes. Genome Biol. Evol. 2014, 7, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Schrader, L.; Kim, J.W.; Ence, D.; Zimin, A.; Klein, A.; Wyschetzki, K.; Weichselgartner, T.; Kemena, C.; Stökl, J.; Schultner, E.; et al. Transposable Element Islands Facilitate Adaptation to Novel Environments in an Invasive Species. Nat. Commun. 2014, 5, 5495. [Google Scholar] [CrossRef] [PubMed]
- Lobachev, K.S.; Stenger, J.E.; Kozyreva, O.G.; Jurka, J.; Gordenin, D.A.; Resnick, M.A. Inverted Alu Repeats Unstable in Yeast Are Excluded from the Human Genome. EMBO J. 2000, 19, 3822–3830. [Google Scholar] [CrossRef] [PubMed]
- Coyne, J.A.; Charlesworth, B. Location of an X-Linked Factor Causing Sterility in Male Hybrids of Drosophila simulans and D. mauritiana. Heredity 1986, 57 Pt 2, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.A.; Wolf, J.B.; Brodie, E.D., III; Wade, M.J. Gene Interactions and the Origin of Species. Epistasis Evol. Process 2000, 94, 197–212. [Google Scholar]
- De Vos, J.M.; Augustijnen, H.; Bätscher, L.; Lucek, K. Speciation through Chromosomal Fusion and Fission in Lepidoptera. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20190539. [Google Scholar] [CrossRef]
- Charlesworth, B. Mutation-Selection Balance and the Evolutionary Advantage of Sex and Recombination. Genet. Res. 1990, 55, 199–221. [Google Scholar] [CrossRef]
- Cutter, A.D.; Payseur, B.A. Genomic Signatures of Selection at Linked Sites: Unifying the Disparity among Species. Nat. Rev. Genet. 2013, 14, 262–274. [Google Scholar] [CrossRef]
- Corbett-Detig, R.B.; Hartl, D.L.; Sackton, T.B. Natural Selection Constrains Neutral Diversity across a Wide Range of Species. PLoS Biol. 2015, 13, e1002112. [Google Scholar] [CrossRef]
- Ortells, M.O. Phylogenetic Analysis of G-Banded Karyotypes among the South American Subterranean Rodents of the Genus Ctenomys (Caviomorpha: Octodontidae), with Special Reference to Chromosomal Evolution and Speciation. Biol. J. Linn. Soc. Lond. 1995, 54, 43–70. [Google Scholar] [CrossRef]
- Pevzner, P.; Tesler, G. Human and Mouse Genomic Sequences Reveal Extensive Breakpoint Reuse in Mammalian Evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 7672–7677. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Herrera, A.; Castresana, J.; Robinson, T.J. Is Mammalian Chromosomal Evolution Driven by Regions of Genome Fragility? Genome Biol. 2006, 7, R115. [Google Scholar] [CrossRef] [PubMed]
- Bulazel, K.V.; Ferreri, G.C.; Eldridge, M.D.B.; O’Neill, R.J. Species-Specific Shifts in Centromere Sequence Composition Are Coincident with Breakpoint Reuse in Karyotypically Divergent Lineages. Genome Biol. 2007, 8, R170. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.J.; Larkin, D.M.; Everts-van der Wind, A.; Bourque, G.; Tesler, G.; Auvil, L.; Beever, J.E.; Chowdhary, B.P.; Galibert, F.; Gatzke, L.; et al. Dynamics of Mammalian Chromosome Evolution Inferred from Multispecies Comparative Maps. Science 2005, 309, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.S.; Carone, D.M.; NISC Comparative Sequencing Program; Green, E.D.; O’Neill, M.J.; O’Neill, R.J. Distinct Retroelement Classes Define Evolutionary Breakpoints Demarcating Sites of Evolutionary Novelty. BMC Genom. 2009, 10, 334. [Google Scholar] [CrossRef] [PubMed]
- Nowak, R.M. Walker’s Mammals of the World; JHU Press: Baltimore, MD, USA, 1999; Volume 1, ISBN 9781421424675. [Google Scholar]
- Trifonov, V.A.; Stanyon, R.; Nesterenko, A.I.; Fu, B.; Perelman, P.L.; O’Brien, P.C.M.; Stone, G.; Rubtsova, N.V.; Houck, M.L.; Robinson, T.J.; et al. Multidirectional Cross-Species Painting Illuminates the History of Karyotypic Evolution in Perissodactyla. Chromosome Res. 2008, 16, 89–107. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, V.A.; Musilova, P.; Kulemsina, A.I. Chromosome Evolution in Perissodactyla. Cytogenet. Genome Res. 2012, 137, 208–217. [Google Scholar] [CrossRef]
- Kumar, S.; Suleski, M.; Craig, J.M.; Kasprowicz, A.E.; Sanderford, M.; Li, M.; Stecher, G.; Hedges, S.B. TimeTree 5: An Expanded Resource for Species Divergence Times. Mol. Biol. Evol. 2022, 39, msac174. [Google Scholar] [CrossRef]
- Piras, F.M.; Nergadze, S.G.; Magnani, E.; Bertoni, L.; Attolini, C.; Khoriauli, L.; Raimondi, E.; Giulotto, E. Uncoupling of Satellite DNA and Centromeric Function in the Genus Equus. PLoS Genet. 2010, 6, e1000845. [Google Scholar] [CrossRef]
- Musilova, P.; Kubickova, S.; Vahala, J.; Rubes, J. Subchromosomal Karyotype Evolution in Equidae. Chromosome Res. 2013, 21, 175–187. [Google Scholar] [CrossRef]
- Steiner, C.C.; Ryder, O.A. Molecular Phylogeny and Evolution of the Perissodactyla. Zool. J. Linn. Soc. 2011, 163, 1289–1303. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, Y.; Bai, D.; Shiraigol, W.; Li, B.; Yang, L.; Wu, J.; Bao, W.; Ren, X.; Jin, B.; et al. Donkey Genome and Insight into the Imprinting of Fast Karyotype Evolution. Sci. Rep. 2015, 5, 14106. [Google Scholar] [CrossRef] [PubMed]
- Piras, F.M.; Nergadze, S.G.; Poletto, V.; Cerutti, F.; Ryder, O.A.; Leeb, T.; Raimondi, E.; Giulotto, E. Phylogeny of Horse Chromosome 5q in the Genus Equus and Centromere Repositioning. Cytogenet. Genome Res. 2009, 126, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Wade, C.M.; Giulotto, E.; Sigurdsson, S.; Zoli, M.; Gnerre, S.; Imsland, F.; Lear, T.L.; Adelson, D.L.; Bailey, E.; Bellone, R.R.; et al. Genome Sequence, Comparative Analysis, and Population Genetics of the Domestic Horse. Science 2009, 326, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Nergadze, S.G.; Piras, F.M.; Gamba, R.; Corbo, M.; Cerutti, F.; McCarter, J.G.W.; Cappelletti, E.; Gozzo, F.; Harman, R.M.; Antczak, D.F.; et al. Birth, Evolution, and Transmission of Satellite-Free Mammalian Centromeric Domains. Genome Res. 2018, 28, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Purgato, S.; Belloni, E.; Piras, F.M.; Zoli, M.; Badiale, C.; Cerutti, F.; Mazzagatti, A.; Perini, G.; Della Valle, G.; Nergadze, S.G.; et al. Centromere Sliding on a Mammalian Chromosome. Chromosoma 2015, 124, 277–287. [Google Scholar] [CrossRef]
- Giulotto, E.; Raimondi, E.; Sullivan, K.F. The Unique DNA Sequences Underlying Equine Centromeres. Prog. Mol. Subcell. Biol. 2017, 56, 337–354. [Google Scholar] [CrossRef]
- Piras, F.M.; Cappelletti, E.; Santagostino, M.; Nergadze, S.G.; Giulotto, E.; Raimondi, E. Molecular Dynamics and Evolution of Centromeres in the Genus Equus. Int. J. Mol. Sci. 2022, 23, 4183. [Google Scholar] [CrossRef]
- Piras, F.M.; Cappelletti, E.; Abdelgadir, W.A.; Salamon, G.; Vignati, S.; Santagostino, M.; Sola, L.; Nergadze, S.G.; Giulotto, E. A Satellite-Free Centromere in Equus Przewalskii Chromosome 10. Int. J. Mol. Sci. 2023, 24, 4134. [Google Scholar] [CrossRef]
- Cerutti, F.; Gamba, R.; Mazzagatti, A.; Piras, F.M.; Cappelletti, E.; Belloni, E.; Nergadze, S.G.; Raimondi, E.; Giulotto, E. The Major Horse Satellite DNA Family Is Associated with Centromere Competence. Mol. Cytogenet. 2016, 9, 35. [Google Scholar] [CrossRef]
- Anglana, M.; Bertoni, L.; Giulotto, E. Cloning of a Polymorphic Sequence from the Nontranscribed Spacer of Horse rDNA. Mamm. Genome 1996, 7, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Nergadze, S.G.; Belloni, E.; Piras, F.M.; Khoriauli, L.; Mazzagatti, A.; Vella, F.; Bensi, M.; Vitelli, V.; Giulotto, E.; Raimondi, E. Discovery and Comparative Analysis of a Novel Satellite, EC137, in Horses and Other Equids. Cytogenet. Genome Res. 2014, 144, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Carbone, L.; Harris, R.A.; Gnerre, S.; Veeramah, K.R.; Lorente-Galdos, B.; Huddleston, J.; Meyer, T.J.; Herrero, J.; Roos, C.; Aken, B.; et al. Gibbon Genome and the Fast Karyotype Evolution of Small Apes. Nature 2014, 513, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, O.; Carbone, L.; Stanyon, R.R.; Marra, A.; Yang, F.; Whelan, C.W.; de Jong, P.J.; Rocchi, M.; Archidiacono, N. A Comprehensive Molecular Cytogenetic Analysis of Chromosome Rearrangements in Gibbons. Genome Res. 2012, 22, 2520–2528. [Google Scholar] [CrossRef]
- Yunis, J.J.; Prakash, O. The Origin of Man: A Chromosomal Pictorial Legacy. Science 1982, 215, 1525–1530. [Google Scholar] [CrossRef]
- Fan, Y.; Linardopoulou, E.; Friedman, C.; Williams, E.; Trask, B.J. Genomic Structure and Evolution of the Ancestral Chromosome Fusion Site in 2q13-2q14.1 and Paralogous Regions on Other Human Chromosomes. Genome Res. 2002, 12, 1651–1662. [Google Scholar] [CrossRef]
- Sangpakdee, W.; Tanomtong, A.; Fan, X.; Pinthong, K.; Weise, A.; Liehr, T. Application of Multicolor Banding Combined with Heterochromatic and Locus-Specific Probes Identify Evolutionary Conserved Breakpoints in Hylobates Pileatus. Mol. Cytogenet. 2016, 9, 17. [Google Scholar] [CrossRef]
- Carbone, L.; Harris, R.A.; Vessere, G.M.; Mootnick, A.R.; Humphray, S.; Rogers, J.; Kim, S.K.; Wall, J.D.; Martin, D.; Jurka, J.; et al. Evolutionary Breakpoints in the Gibbon Suggest Association between Cytosine Methylation and Karyotype Evolution. PLoS Genet. 2009, 5, e1000538. [Google Scholar] [CrossRef]
- Carbone, L.; Harris, R.A.; Mootnick, A.R.; Milosavljevic, A.; Martin, D.I.K.; Rocchi, M.; Capozzi, O.; Archidiacono, N.; Konkel, M.K.; Walker, J.A.; et al. Centromere Remodeling in Hoolock Leuconedys (Hylobatidae) by a New Transposable Element Unique to the Gibbons. Genome Biol. Evol. 2012, 4, 648–658. [Google Scholar] [CrossRef]
- Okhovat, M.; Nevonen, K.A.; Davis, B.A.; Michener, P.; Ward, S.; Milhaven, M.; Harshman, L.; Sohota, A.; Fernandes, J.D.; Salama, S.R.; et al. Co-Option of the Lineage-Specific LAVA Retrotransposon in the Gibbon Genome. Proc. Natl. Acad. Sci. USA 2020, 117, 19328–19338. [Google Scholar] [CrossRef]
- Meyer, T.J.; Held, U.; Nevonen, K.A.; Klawitter, S.; Pirzer, T.; Carbone, L.; Schumann, G.G. The Flow of the Gibbon LAVA Element Is Facilitated by the LINE-1 Retrotransposition Machinery. Genome Biol. Evol. 2016, 8, 3209–3225. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, I.; Kazakov, A.; Tumeneva, I.; Shepelev, V.; Yurov, Y. Alpha-Satellite DNA of Primates: Old and New Families. Chromosoma 2001, 110, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Pironon, N.; Puechberty, J.; Roizès, G. Molecular and Evolutionary Characteristics of the Fraction of Human Alpha Satellite DNA Associated with CENP-A at the Centromeres of Chromosomes 1, 5, 19, and 21. BMC Genom. 2010, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Altemose, N.; Logsdon, G.A.; Bzikadze, A.V.; Sidhwani, P.; Langley, S.A.; Caldas, G.V.; Hoyt, S.J.; Uralsky, L.; Ryabov, F.D.; Shew, C.J.; et al. Complete Genomic and Epigenetic Maps of Human Centromeres. Science 2022, 376, eabl4178. [Google Scholar] [CrossRef] [PubMed]
- Koga, A.; Hirai, Y.; Hara, T.; Hirai, H. Repetitive Sequences Originating from the Centromere Constitute Large-Scale Heterochromatin in the Telomere Region in the Siamang, a Small Ape. Heredity 2012, 109, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Baicharoen, S.; Miyabe-Nishiwaki, T.; Arsaithamkul, V.; Hirai, Y.; Duangsa-ard, K.; Siriaroonrat, B.; Domae, H.; Srikulnath, K.; Koga, A.; Hirai, H. Locational Diversity of Alpha Satellite DNA and Intergeneric Hybridization Aspects in the Nomascus and Hylobates Genera of Small Apes. PLoS ONE 2014, 9, e109151. [Google Scholar] [CrossRef] [PubMed]
- Trizzino, M.; Park, Y.; Holsbach-Beltrame, M.; Aracena, K.; Mika, K.; Caliskan, M.; Perry, G.H.; Lynch, V.J.; Brown, C.D. Transposable Elements Are the Primary Source of Novelty in Primate Gene Regulation. Genome Res. 2017, 27, 1623–1633. [Google Scholar] [CrossRef]
- Hollister, J.D.; Gaut, B.S. Epigenetic Silencing of Transposable Elements: A Trade-off between Reduced Transposition and Deleterious Effects on Neighboring Gene Expression. Genome Res. 2009, 19, 1419–1428. [Google Scholar] [CrossRef]
- Brown, J.D.; O’Neill, R.J. The Mysteries of Chromosome Evolution in Gibbons: Methylation Is a Prime Suspect. PLoS Genet. 2009, 5, e1000501. [Google Scholar] [CrossRef]
- O’Neill, R.J.; O’Neill, M.J.; Graves, J.A. Undermethylation Associated with Retroelement Activation and Chromosome Remodelling in an Interspecific Mammalian Hybrid. Nature 1998, 393, 68–72. [Google Scholar] [CrossRef]
- Ferreri, G.C.; Brown, J.D.; Obergfell, C.; Jue, N.; Finn, C.E.; O’Neill, M.J.; O’Neill, R.J. Recent Amplification of the Kangaroo Endogenous Retrovirus, KERV, Limited to the Centromere. J. Virol. 2011, 85, 4761–4771. [Google Scholar] [CrossRef] [PubMed]
- Koina, E.; Fong, J.; Graves, J.A.M. Marsupial and Monotreme Genomes. Genome Dyn. 2006, 2, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Meredith, R.W.; Westerman, M.; Case, J.A.; Springer, M.S. A Phylogeny and Timescale for Marsupial Evolution Based on Sequences for Five Nuclear Genes. J. Mamm. Evol. 2008, 15, 1–36. [Google Scholar] [CrossRef]
- O’Neill, R.J.; Eldridge, M.D.; Graves, J.A. Chromosome Heterozygosity and de Novo Chromosome Rearrangements in Mammalian Interspecies Hybrids. Mamm. Genome 2001, 12, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, C.J.; Bulazel, K.V.; Ferreri, G.C.; Schroeder-Reiter, E.; Wanner, G.; Rens, W.; Obergfell, C.; Eldridge, M.D.B.; O’Neill, R.J. Genomic Instability within Centromeres of Interspecific Marsupial Hybrids. Genetics 2007, 177, 2507–2517. [Google Scholar] [CrossRef]
- Rens, W.; Ferguson-Smith, M. The Conserved Marsupial Karyotype: Chromosome Painting and Evolution. In Marsupial Genetics and Genomics; Springer: Berlin/Heidelberg, Germany, 2010; pp. 37–53. [Google Scholar]
- Rens, W.; O’Brien, P.C.M.; Fairclough, H.; Harman, L.; Graves, J.A.M.; Ferguson-Smith, M.A. Reversal and Convergence in Marsupial Chromosome Evolution. Cytogenet. Genome Res. 2003, 102, 282–290. [Google Scholar] [CrossRef]
- Deakin, J. Chromosome Evolution in Marsupials. Genes 2018, 9, 72. [Google Scholar] [CrossRef]
- Deakin, J.E.; Delbridge, M.L.; Koina, E.; Harley, N.; Alsop, A.E.; Wang, C.; Patel, V.S.; Graves, J.A.M. Reconstruction of the Ancestral Marsupial Karyotype from Comparative Gene Maps. BMC Evol. Biol. 2013, 13, 258. [Google Scholar] [CrossRef]
- O’Neill, R.J.; Eldridge, M.D.B.; Metcalfe, C.J. Centromere Dynamics and Chromosome Evolution in Marsupials. J. Hered. 2004, 95, 375–381. [Google Scholar] [CrossRef]
- Ohno, S. Sex Chromosomes and Sex-Linked Genes; Springer: Berlin/Heidelberg, Germany, 1966; p. 192. [Google Scholar]
- Sharman, G.B.; McINTOSH, A.J.; Barber, H.N. Multiple Sex Chromosomes in the Marsupials. Nature 1950, 166, 996. [Google Scholar] [CrossRef]
- Toder, R.; O’Neill, R.J.; Wienberg, J.; O’Brien, P.C.; Voullaire, L.; Marshall-Graves, J.A. Comparative Chromosome Painting between Two Marsupials: Origins of an XX/XY1Y2 Sex Chromosome System. Mamm. Genome 1997, 8, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.W.; Hjelmen, C.E.; Blackmon, H. The Probability of Fusions Joining Sex Chromosomes and Autosomes. Biol. Lett. 2020, 16, 20200648. [Google Scholar] [CrossRef] [PubMed]
- Amar, L.C.; Dandolo, L.; Hanauer, A.; Cook, A.R.; Arnaud, D.; Mandel, J.L.; Avner, P. Conservation and Reorganization of Loci on the Mammalian X Chromosome: A Molecular Framework for the Identification of Homologous Subchromosomal Regions in Man and Mouse. Genomics 1988, 2, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial Sequencing and Comparative Analysis of the Mouse Genome. Nature 2002, 420, 520–562. [Google Scholar] [CrossRef] [PubMed]
- Sandstedt, S.A.; Tucker, P.K. Evolutionary Strata on the Mouse X Chromosome Correspond to Strata on the Human X Chromosome. Genome Res. 2004, 14, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Deakin, J.E.; Koina, E.; Waters, P.D.; Doherty, R.; Patel, V.S.; Delbridge, M.L.; Dobson, B.; Fong, J.; Hu, Y.; van den Hurk, C.; et al. Physical Map of Two Tammar Wallaby Chromosomes: A Strategy for Mapping in Non-Model Mammals. Chromosome Res. 2008, 16, 1159–1175. [Google Scholar] [CrossRef] [PubMed]
- Deakin, J.E. Marsupial Genome Sequences: Providing Insight into Evolution and Disease. Scientifica 2012, 2012, 1–22. [Google Scholar] [CrossRef]
- Deakin, J.E. Marsupial X Chromosome Inactivation: Past, Present and Future. Aust. J. Zool. 2013, 61, 13. [Google Scholar] [CrossRef]
- Carr, D.H.; Singh, R.P.; Miller, I.R.; McGeer, P.L. The Chromosome Complement of the Pacific Killer Whale (Orcinus rectipinna). Mammal Chromosomes Newslett 1966, 22, 208. [Google Scholar]
- Arnason, U. The Karyotype of the Fin Whale. Hereditas 1969, 62, 273–284. [Google Scholar] [CrossRef]
- Arnason, U. Karyotypes of a Male Sperm Whale (Physeter catodon L.) and a Female Sei Whale (Balaenoptera borealis Less.). Hereditas 1970, 64, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Arnason, U.; Benirschke, K.; Mead, J.G.; Nichols, W.W. Banded Karyotypes of Three Whales: Mesoplodon europaeus, M. carlhubbsi and Balaenoptera acutorostrata. Hereditas 1977, 87, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Heinzelmann, L.; Chagastelles, P.C.; Danilewicz, D.; Chies, J.A.B.; Andrades-Miranda, J. The Karyotype of Franciscana Dolphin (Pontoporia blainvillei). J. Hered. 2009, 100, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Kulemzina, A.I.; Proskuryakova, A.A.; Beklemisheva, V.R.; Lemskaya, N.A.; Perelman, P.L.; Graphodatsky, A.S. Comparative Chromosome Map and Heterochromatin Features of the Gray Whale Karyotype (Cetacea). Cytogenet. Genome Res. 2016, 148, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Mudd, A.B.; Bredeson, J.V.; Baum, R.; Hockemeyer, D.; Rokhsar, D.S. Analysis of Muntjac Deer Genome and Chromatin Architecture Reveals Rapid Karyotype Evolution. Commun. Biol. 2020, 3, 480. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; O’Brien, P.C.; Wienberg, J.; Ferguson-Smith, M.A. A Reappraisal of the Tandem Fusion Theory of Karyotype Evolution in Indian Muntjac Using Chromosome Painting. Chromosome Res. 1997, 5, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Chi, J.; Nie, W.; Wang, J.; Yang, F. Phylogenomics of Several Deer Species Revealed by Comparative Chromosome Painting with Chinese Muntjac Paints. Genetica 2006, 127, 25–33. [Google Scholar] [CrossRef]
- Wang, W.; Lan, H. Rapid and Parallel Chromosomal Number Reductions in Muntjac Deer Inferred from Mitochondrial DNA Phylogeny. Mol. Biol. Evol. 2000, 17, 1326–1333. [Google Scholar] [CrossRef]
- Wurster, D.H.; Benirschke, K. Indian Muntjac, Muntiacus Muntjak: A Deer with a Low Diploid Chromosome Number. Science 1970, 168, 1364–1366. [Google Scholar] [CrossRef]
- Yang, F.; Carter, N.P.; Shi, L.; Ferguson-Smith, M.A. A Comparative Study of Karyotypes of Muntjacs by Chromosome Painting. Chromosoma 1995, 103, 642–652. [Google Scholar] [CrossRef]
- Frönicke, L.; Scherthan, H. Zoo-Fluorescence in Situ Hybridization Analysis of Human and Indian Muntjac Karyotypes (Muntiacus Muntjak Vaginalis) Reveals Satellite DNA Clusters at the Margins of Conserved Syntenic Segments. Chromosome Res. 1997, 5, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Tsipouri, V.; Schueler, M.G.; Hu, S.; NISC Comparative Sequencing Program; Dutra, A.; Pak, E.; Riethman, H.; Green, E.D. Comparative Sequence Analyses Reveal Sites of Ancestral Chromosomal Fusions in the Indian Muntjac Genome. Genome Biol. 2008, 9, R155. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.X.; Huang, L.; Nie, W.; Wang, J.; Su, B.; Yang, F. Defining the Orientation of the Tandem Fusions That Occurred during the Evolution of Indian Muntjac Chromosomes by BAC Mapping. Chromosoma 2005, 114, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Chi, J.; Wang, J.; Nie, W.; Su, W.; Yang, F. High-Density Comparative BAC Mapping in the Black Muntjac (Muntiacus Crinifrons): Molecular Cytogenetic Dissection of the Origin of MCR 1p+4 in the X1X2Y1Y2Y3 Sex Chromosome System. Genomics 2006, 87, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.J.; Stanyon, R.; O’Brien, S.J. Evolution of Mammalian Genome Organization Inferred from Comparative Gene Mapping. Genome Biol. 2001, 2, REVIEWS0005. [Google Scholar] [CrossRef]
- Huang, L.; Wang, J.; Nie, W.; Su, W.; Yang, F. Tandem Chromosome Fusions in Karyotypic Evolution of Muntiacus: Evidence from M. Feae and M. Gongshanensis. Chromosome Res. 2006, 14, 637–647. [Google Scholar] [CrossRef]
- McArthur, E.; Capra, J.A. Topologically Associating Domain Boundaries That Are Stable across Diverse Cell Types Are Evolutionarily Constrained and Enriched for Heritability. Am. J. Hum. Genet. 2021, 108, 269–283. [Google Scholar] [CrossRef]
- Krefting, J.; Andrade-Navarro, M.A.; Ibn-Salem, J. Evolutionary Stability of Topologically Associating Domains Is Associated with Conserved Gene Regulation. BMC Biol. 2018, 16, 87. [Google Scholar] [CrossRef]
- Lazar, N.H.; Nevonen, K.A.; O’Connell, B.; McCann, C.; O’Neill, R.J.; Green, R.E.; Meyer, T.J.; Okhovat, M.; Carbone, L. Epigenetic Maintenance of Topological Domains in the Highly Rearranged Gibbon Genome. Genome Res. 2018, 28, 983–997. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The Complete Sequence of a Human Genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Rautiainen, M.; Nurk, S.; Walenz, B.P.; Logsdon, G.A.; Porubsky, D.; Rhie, A.; Eichler, E.E.; Phillippy, A.M.; Koren, S. Verkko: Telomere-to-Telomere Assembly of Diploid Chromosomes. bioRxiv 2022, 2022.06.24.497523. [Google Scholar] [CrossRef] [PubMed]
- Hoyt, S.J.; Storer, J.M.; Hartley, G.A.; Grady, P.G.S.; Gershman, A.; de Lima, L.G.; Limouse, C.; Halabian, R.; Wojenski, L.; Rodriguez, M.; et al. From Telomere to Telomere: The Transcriptional and Epigenetic State of Human Repeat Elements. Science 2022, 376, eabk3112. [Google Scholar] [CrossRef] [PubMed]
- T2T Consortium. Available online: https://sites.google.com/ucsc.edu/t2tworkinggroup (accessed on 28 December 2023).
- GenomeArk. Available online: https://www.genomeark.org/ (accessed on 28 December 2023).
- Lewin, H.A. Earth BioGenome Project: Sequencing Life for the Future of Life. Proc. Natl. Acad. Sci. USA 2018, 115, 4325–4333. [Google Scholar] [CrossRef] [PubMed]
- Vertebrate Genomes Project. Available online: https://vertebrategenomesproject.org/ (accessed on 28 December 2023).
- WHOI to Co-Lead Deep-Ocean Genomes Project. Available online: https://www.whoi.edu/press-room/news-release/earth-biogenome-project/ (accessed on 30 November 2023).
- Bright, M. Darwin’s Tree of Life; Wayland: London, UK, 2022; ISBN 9781526306364. [Google Scholar]
- Oz Mammals Genomics. Available online: https://ozmammalsgenomics.com/ (accessed on 30 November 2023).


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brannan, E.O.; Hartley, G.A.; O’Neill, R.J. Mechanisms of Rapid Karyotype Evolution in Mammals. Genes 2024, 15, 62. https://doi.org/10.3390/genes15010062
Brannan EO, Hartley GA, O’Neill RJ. Mechanisms of Rapid Karyotype Evolution in Mammals. Genes. 2024; 15(1):62. https://doi.org/10.3390/genes15010062
Chicago/Turabian StyleBrannan, Emry O., Gabrielle A. Hartley, and Rachel J. O’Neill. 2024. "Mechanisms of Rapid Karyotype Evolution in Mammals" Genes 15, no. 1: 62. https://doi.org/10.3390/genes15010062
APA StyleBrannan, E. O., Hartley, G. A., & O’Neill, R. J. (2024). Mechanisms of Rapid Karyotype Evolution in Mammals. Genes, 15(1), 62. https://doi.org/10.3390/genes15010062