
Genome-Wide Association Studies of Hair Whorl in Pigs
 ,
, 

Abstract
1. Introduction
2. Materials and Methods
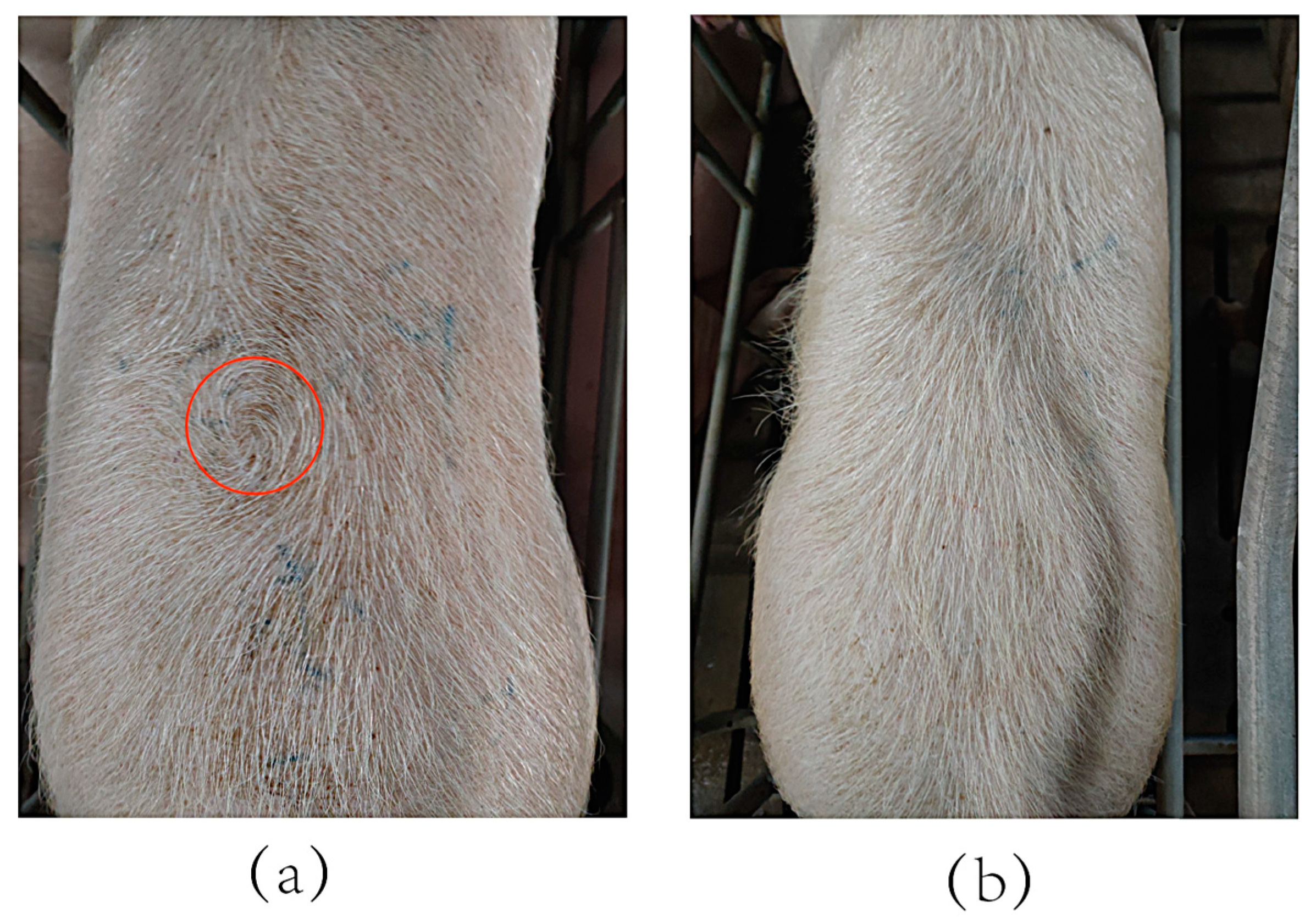
2.1. Study Population and Phenotypic Determination
2.2. Performance Measurement and Slaughter Measurement
2.3. Genotyping and Quality Control
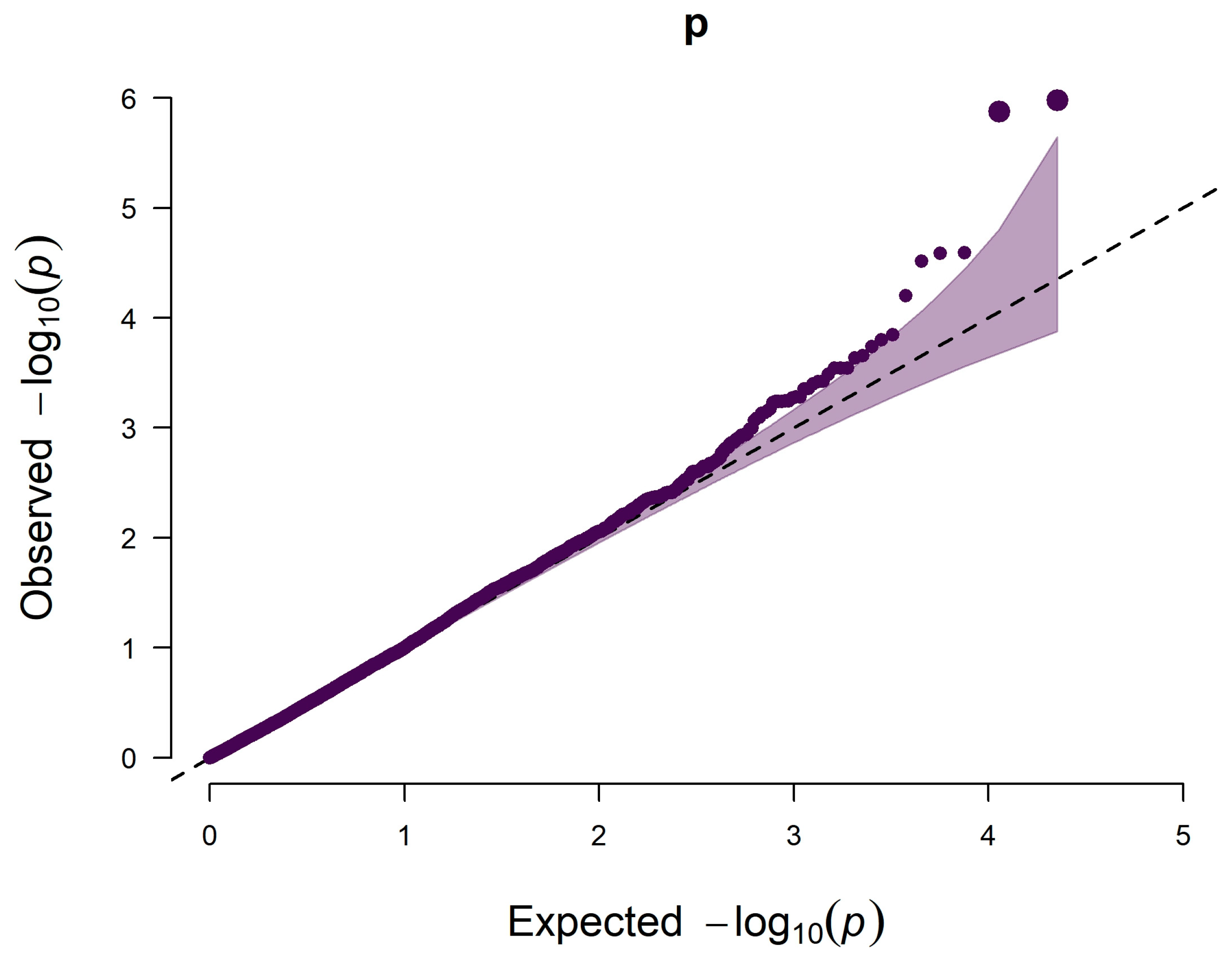
2.4. Genome-Wide Association Studies
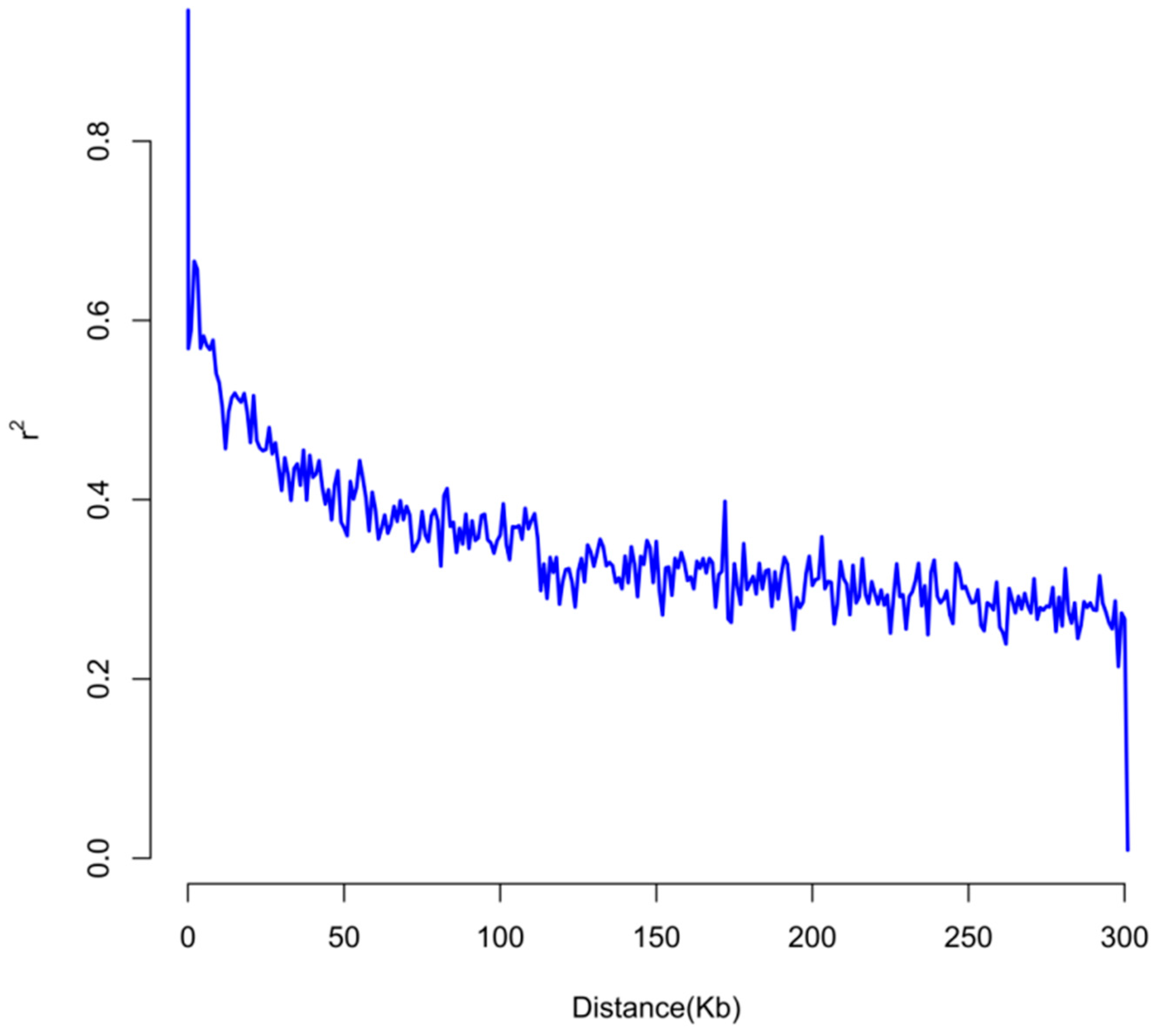
2.5. Linkage Disequilibrium Analysis
2.6. Analysis of Copy Number Variation
2.7. Identification of Candidate Genes
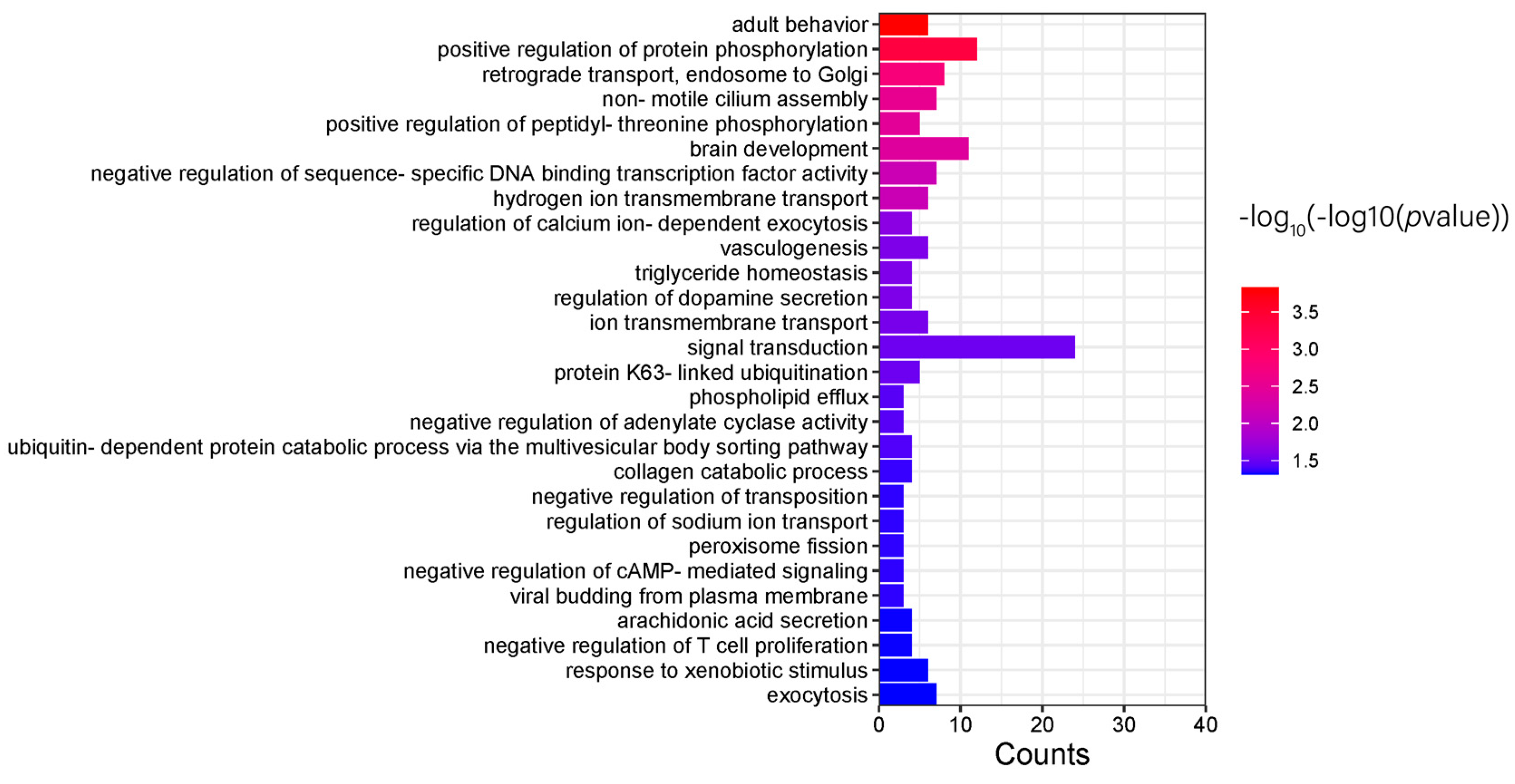
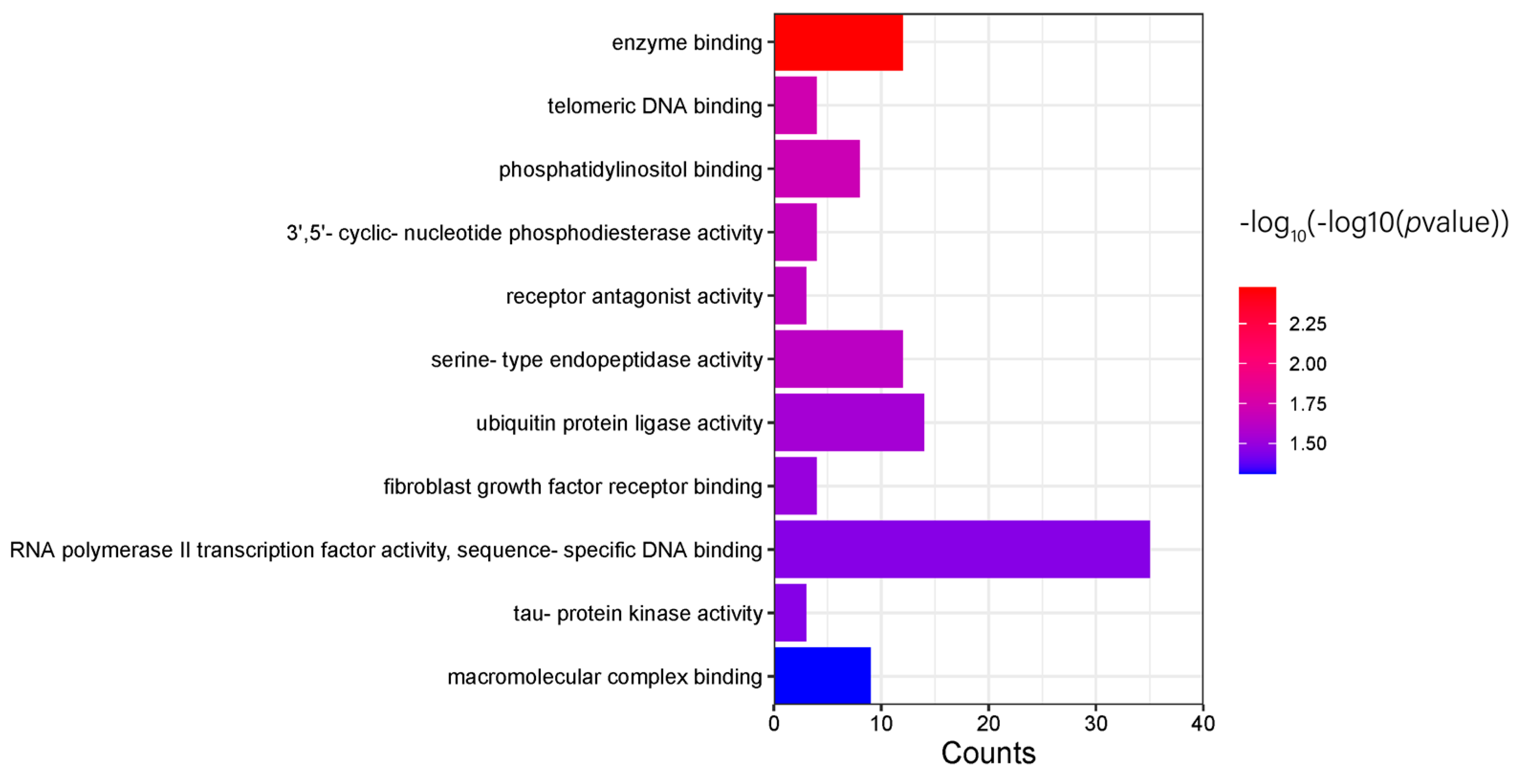
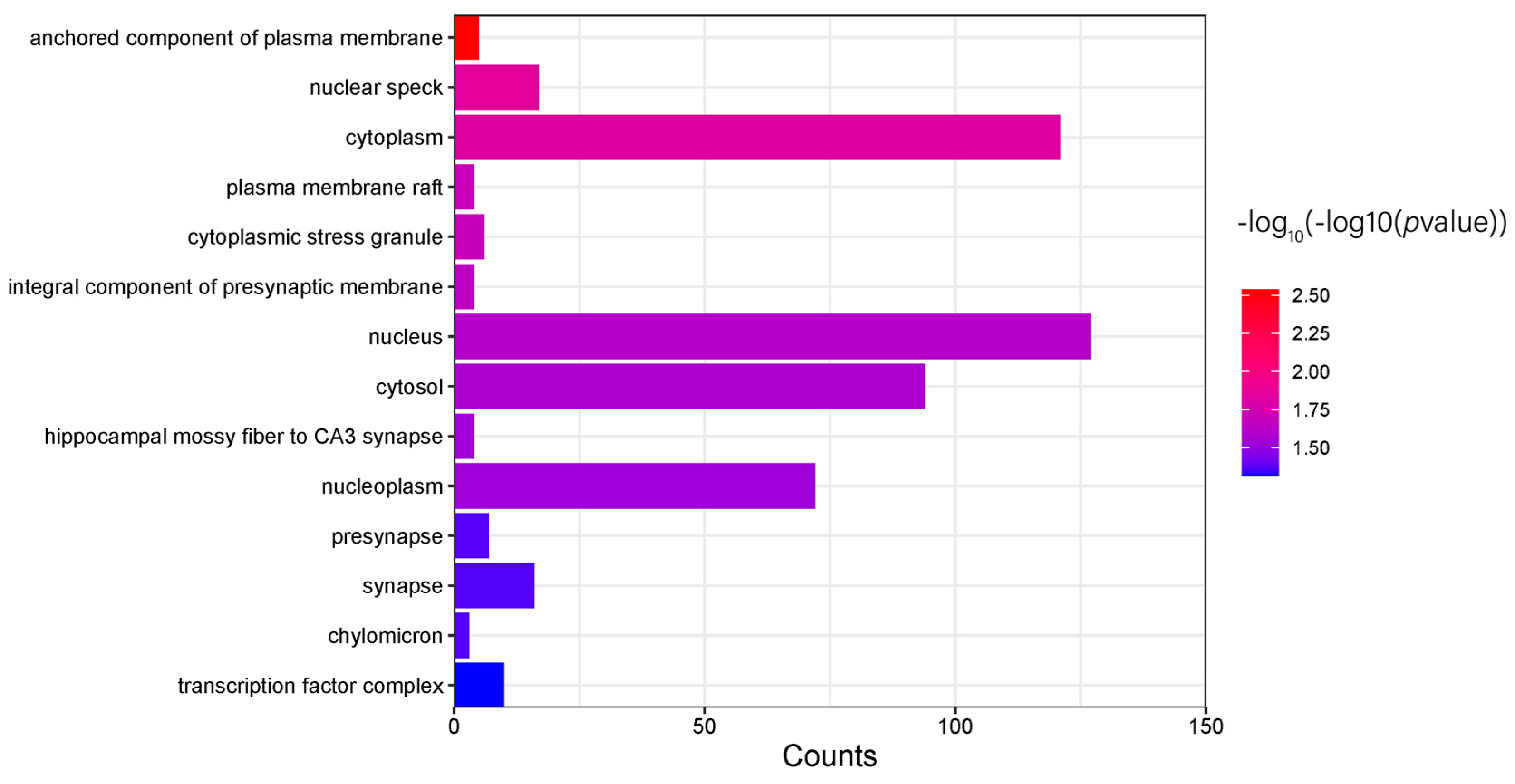
2.8. Tional Enrichment Analysis
3. Results
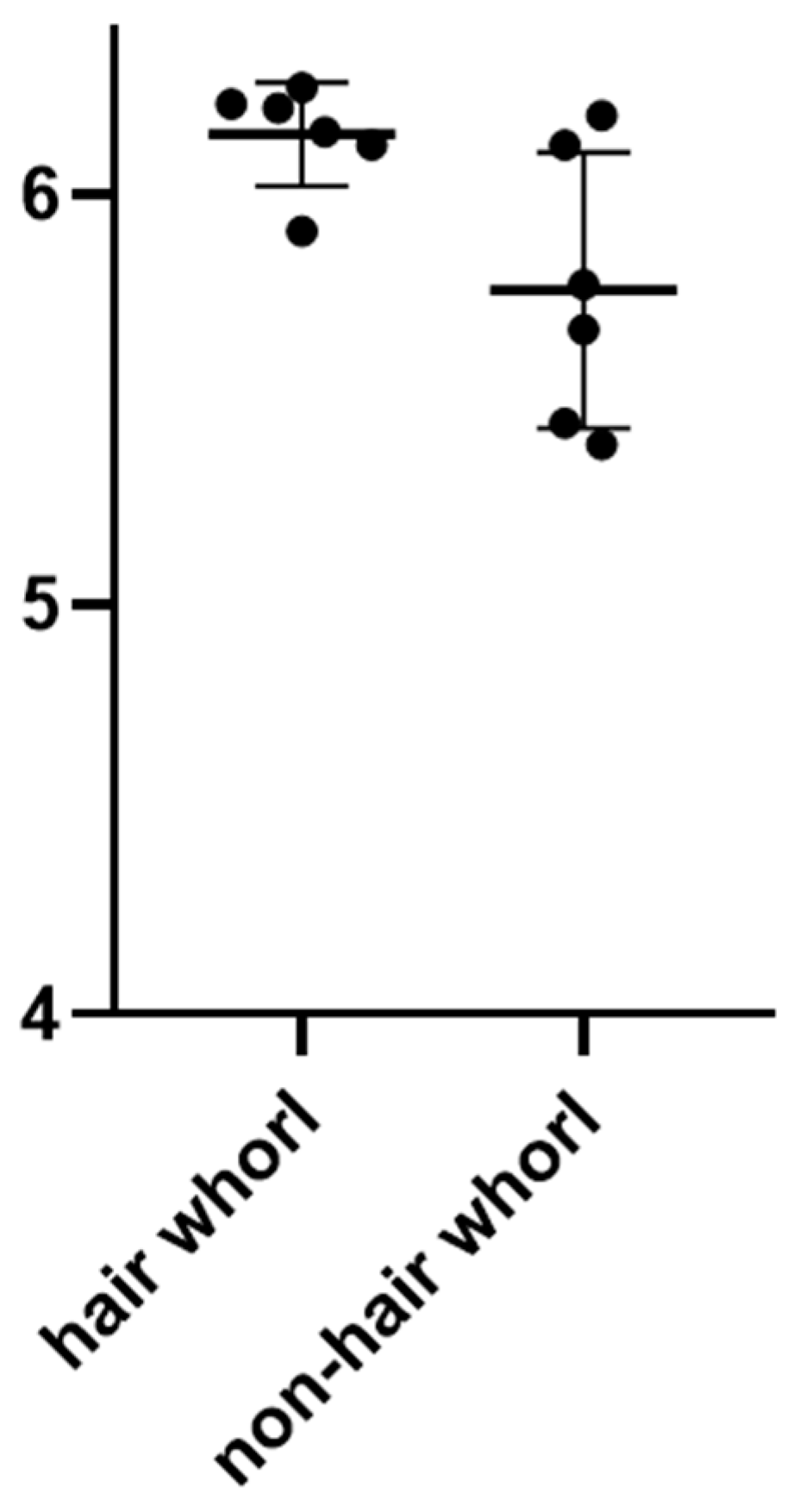
3.1. Performance Measure
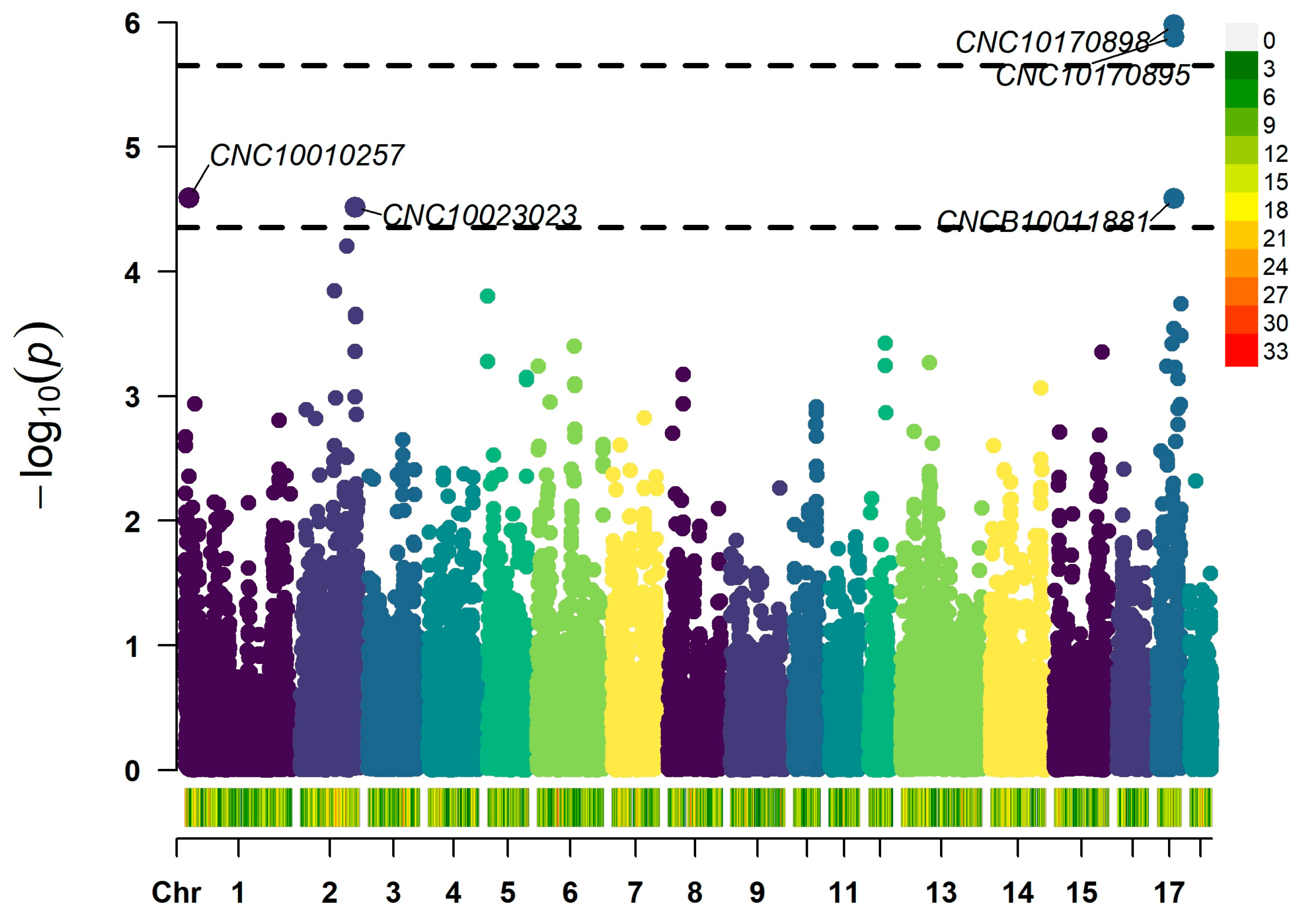
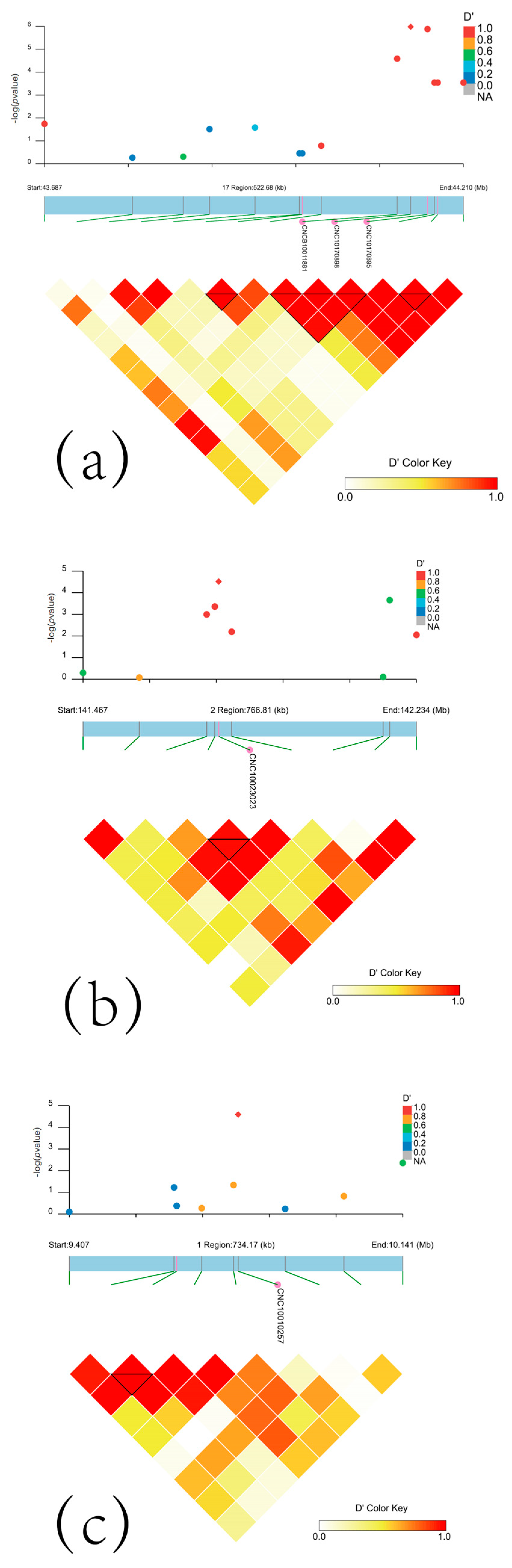
3.2. The Significant SNPs for Hair Whorl Trait
3.3. Linkage Disequilibrium Results
3.4. Results of Copy Number Variation Analysis
3.5. Candidate Gene and Functional Annotation
3.5.1. GWAS Results
3.5.2. CNV Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhuang, Z.; Wu, J.; Xu, C.; Ruan, D.; Qiu, Y.; Zhou, S.; Ding, R.; Quan, J.; Yang, M.; Zheng, E.; et al. The Genetic Architecture of Meat Quality Traits in a Crossbred Commercial Pig Population. Foods 2022, 11, 3143. [Google Scholar] [CrossRef] [PubMed]
- Grindflek, E.; Hansen, M.H.S.; Lien, S.; van Son, M. Genome-wide association study reveals a QTL and strong candidate genes for umbilical hernia in pigs on SSC14. BMC Genom. 2018, 19, 412. [Google Scholar] [CrossRef] [PubMed]
- Anil, S.S.; Anil, L.; Deen, J. Effect of lameness on sow longevity. J. Am. Vet. Med. Assoc. 2009, 235, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Jiang, Y.; Zhang, H.; Mei, M.; Song, H.; Ma, X.; Jiang, L.; Yu, Z.; Zhang, Q.; Ding, X. A mutation in MAP2 is associated with prenatal hair follicle density. FASEB J. 2019, 33, 14479–14490. [Google Scholar] [CrossRef]
- Jiang, Y.; Zou, Q.; Liu, B.; Li, S.; Wang, Y.; Liu, T.; Ding, X. Atlas of Prenatal Hair Follicle Morphogenesis Using the Pig as a Model System. Front. Cell Dev. Biol. 2021, 9, 721979. [Google Scholar] [CrossRef]
- Hausman, G.J.; Martin, R.J. The development of adipocytes located around hair follicles in the fetal pig. J. Anim. Sci. 1982, 54, 1286–1296. [Google Scholar] [CrossRef]
- Encina, A.; Ligero, M.; Sánchez-Guerrero, M.J.; Rodríguez-Sainz de Los Terreros, A.; Bartolomé, E.; Valera, M. Phenotypic and Genetic Study of the Presence of Hair Whorls in Pura Raza Español Horses. Animals 2023, 13, 2943. [Google Scholar] [CrossRef]
- Lima, D.; da Cruz, V.A.R.; Pereira, G.L.; Curi, R.A.; Costa, R.B.; de Camargo, G.M.F. Genomic Regions Associated with the Position and Number of Hair Whorls in Horses. Animals 2021, 11, 2925. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Shi, L.; Zhang, P.; Li, Y.; Li, M.; Tian, J.; Wang, L.; Zhao, F. GWAS of Reproductive Traits in Large White Pigs on Chip and Imputed Whole-Genome Sequencing Data. Int. J. Mol. Sci. 2022, 23, 3338. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Gong, H.; Cui, L.; Zhang, W.; Ma, J.; Chen, C.; Ai, H.; Xiao, S.; Huang, L.; et al. Genetic correlation of fatty acid composition with growth, carcass, fat deposition and meat quality traits based on GWAS data in six pig populations. Meat Sci. 2019, 150, 47–55. [Google Scholar] [CrossRef]
- Xu, P.; Ni, L.; Tao, Y.; Ma, Z.; Hu, T.; Zhao, X.; Yu, Z.; Lu, C.; Zhao, X.; Ren, J. Genome-wide association study for growth and fatness traits in Chinese Sujiang pigs. Anim. Genet. 2020, 51, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Yan, G.; Chen, D.; Xiao, S.; Gao, J.; Zhang, Z. An association study using imputed whole-genome sequence data identifies novel significant loci for growth-related traits in a Duroc x Erhualian F(2) population. J. Anim. Breed. Genet. 2019, 136, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, X.; Tan, Z.; Xing, K.; Yang, T.; Wang, Y.; Sun, D.; Wang, C. Genome-wide association study for reproductive traits in a Large White pig population. Anim. Genet. 2018, 49, 127–131. [Google Scholar] [CrossRef]
- Verardo, L.L.; Silva, F.F.; Lopes, M.S.; Madsen, O.; Bastiaansen, J.W.; Knol, E.F.; Kelly, M.; Varona, L.; Lopes, P.S.; Guimaraes, S.E. Revealing new candidate genes for reproductive traits in pigs: Combining Bayesian GWAS and functional pathways. Genet. Sel. Evol. 2016, 48, 9. [Google Scholar] [CrossRef]
- Bakoev, S.; Getmantseva, L.; Bakoev, F.; Kolosova, M.; Gabova, V.; Kolosov, A.; Kostyunina, O. Survey of SNPs Associated with Total Number Born and Total Number Born Alive in Pig. Genes 2020, 11, 491. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Gong, H.; Cui, L.; Ma, J.; Chen, C.; Ai, H.; Xiao, S.; Huang, L.; Yang, B. Landscape of Loci and Candidate Genes for Muscle Fatty Acid Composition in Pigs Revealed by Multiple Population Association Analysis. Front. Genet. 2019, 10, 1067. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Li, M.; Sun, H.; Chen, Q.; Yan, D.; Dong, X.; Pan, Y.; Lu, S. Genome-wide association study reveals genetic loci and candidate genes for meat quality traits in a four-way crossbred pig population. Front. Genet. 2023, 14, 1001352. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, X.; Zheng, Z.; Ma, T.; Liu, Y.; Long, H.; Cheng, H.; Fang, M.; Gong, J.; Li, X.; et al. Genome-wide analysis of expression QTL (eQTL) and allele-specific expression (ASE) in pig muscle identifies candidate genes for meat quality traits. Genet. Sel. Evol. 2020, 52, 59. [Google Scholar] [CrossRef]
- Walker, L.R.; Jobman, E.E.; Sutton, K.M.; Wittler, J.; Johnson, R.K.; Ciobanu, D.C. Genome-wide association analysis for porcine reproductive and respiratory syndrome virus susceptibility traits in two genetic populations of pigs1. J. Anim. Sci. 2019, 97, 3253–3261. [Google Scholar] [CrossRef]
- Uemoto, Y.; Ichinoseki, K.; Matsumoto, T.; Oka, N.; Takamori, H.; Kadowaki, H.; Kojima-Shibata, C.; Suzuki, E.; Okamura, T.; Aso, H.; et al. Genome-wide association studies for production, respiratory disease, and immune-related traits in Landrace pigs. Sci. Rep. 2021, 11, 15823. [Google Scholar] [CrossRef]
- Oyelami, F.O.; Zhao, Q.; Xu, Z.; Zhang, Z.; Sun, H.; Zhang, Z.; Ma, P.; Wang, Q.; Pan, Y. Haplotype Block Analysis Reveals Candidate Genes and QTLs for Meat Quality and Disease Resistance in Chinese Jiangquhai Pig Breed. Front. Genet. 2020, 11, 752. [Google Scholar] [CrossRef] [PubMed]
- Qian, R.; Xie, F.; Zhang, W.; Kong, J.; Zhou, X.; Wang, C.; Li, X. Genome-wide detection of CNV regions between Anqing six-end-white and Duroc pigs. Mol. Cytogenet. 2023, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- Marklund, S.; Kijas, J.; Rodriguez-Martinez, H.; Rönnstrand, L.; Funa, K.; Moller, M.; Lange, D.; Edfors-Lilja, I.; Andersson, L. Molecular basis for the dominant white phenotype in the domestic pig. Genome Res. 1998, 8, 826–833. [Google Scholar] [CrossRef]
- Zheng, X.; Zhao, P.; Yang, K.; Ning, C.; Wang, H.; Zhou, L.; Liu, J. CNV analysis of Meishan pig by next-generation sequencing and effects of AHR gene CNV on pig reproductive traits. J. Anim. Sci. Biotechnol. 2020, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Marees, A.T.; de Kluiver, H.; Stringer, S.; Vorspan, F.; Curis, E.; Marie-Claire, C.; Derks, E.M. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int. J. Methods Psychiatr. Res. 2018, 27, e1608. [Google Scholar] [CrossRef]
- Wang, D.; Tang, H.; Liu, J.F.; Xu, S.; Zhang, Q.; Ning, C. Rapid epistatic mixed-model association studies by controlling multiple polygenic effects. Bioinformatics 2020, 36, 4833–4837. [Google Scholar] [CrossRef]
- RC Team. R: A Language and Environment for Statistical Computing; RC Team: Vienna, Austria, 2023. [Google Scholar]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Dong, S.-S.; He, W.-M.; Ji, J.-J.; Zhang, C.; Guo, Y.; Yang, T.-L. LDBlockShow: A fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief. Bioinform. 2020, 22, bbaa227. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.; Hakonarson, H.; Bucan, M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Smedley, D.; Haider, S.; Ballester, B.; Holland, R.; London, D.; Thorisson, G.; Kasprzyk, A. BioMart--biological queries made easy. BMC Genom. 2009, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.; Machado, P.C.; Pinto, L.F.B.; Silva, M.R.; Schenkel, F.S.; Brito, L.F.; Pedrosa, V.B. Genome-wide association study and pathway analysis for fat deposition traits in Nellore cattle raised in pasture-based systems. J. Anim. Breed. Genet. 2021, 138, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Adapala, N.S.; Kim, H.K. Comprehensive Genome-Wide Transcriptomic Analysis of Immature Articular Cartilage following Ischemic Osteonecrosis of the Femoral Head in Piglets. PLoS ONE 2016, 11, e0153174. [Google Scholar] [CrossRef]
- Simonetti, O.; Lucarini, G.; Bernardini, M.L.; Simoncini, C.; Biagini, G.; Offidani, A. Expression of vascular endothelial growth factor, apoptosis inhibitors (survivin and p16) and CCL27 in alopecia areata before and after diphencyprone treatment: An immunohistochemical study. Br. J. Dermatol. 2004, 150, 940–948. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell. Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef]
- Green, M.R.; Couchman, J.R. Distribution of epidermal growth factor receptors in rat tissues during embryonic skin development, hair formation, and the adult hair growth cycle. J. Investig. Dermatol. 1984, 83, 118–123. [Google Scholar] [CrossRef]
- Wynn, P.C.; Brown, G.; Moore, G.P. Characterization and distribution of epidermal growth factor receptors in the skin and wool follicles of the sheep fetus during development. Domest. Anim. Endocrinol. 1995, 12, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Nanba, D.; Toki, F.; Barrandon, Y.; Higashiyama, S. Recent advances in the epidermal growth factor receptor/ligand system biology on skin homeostasis and keratinocyte stem cell regulation. J. Dermatol. Sci. 2013, 72, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Klufa, J.; Bauer, T.; Hanson, B.; Herbold, C.; Starkl, P.; Lichtenberger, B.; Srutkova, D.; Schulz, D.; Vujic, I.; Mohr, T.; et al. Hair eruption initiates and commensal skin microbiota aggravate adverse events of anti-EGFR therapy. Sci. Transl. Med. 2019, 11, eaax2693. [Google Scholar] [CrossRef] [PubMed]
- Banks, R.W.; Cahusac, P.M.; Graca, A.; Kain, N.; Shenton, F.; Singh, P.; Njå, A.; Simon, A.; Watson, S.; Slater, C.R.; et al. Glutamatergic modulation of synaptic-like vesicle recycling in mechanosensory lanceolate nerve terminals of mammalian hair follicles. J. Physiol. 2013, 591, 2523–2540. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Individuals | Min | Mean | Max | SD |
|---|---|---|---|---|---|
| Number of Live litter size | 2096 | 1 | 12.27 | 21 | 3.57 |
| Number of Total litter size | 2097 | 1 | 12.88 | 21 | 3.74 |
| Number of healthy litters | 2096 | 1 | 10.93 | 18 | 3.09 |
| Birth weight | 2067 | 0.6 | 1.25 | 2.2 | 0.25 |
| Number of left teats | 2096 | 5 | 7.04 | 9 | 0.61 |
| Number of right teats | 2096 | 5 | 6.91 | 9 | 0.58 |
| Corrected 100 kg weight of age | 1851 | 129.89 | 159.79 | 232.79 | 18.33 |
| Corrected 100 kg backfat | 1851 | 5.11 | 12.15 | 24.39 | 2.81 |
| Corrected FCR | 659 | 1.41 | 1.85 | 3.17 | 0.30 |
| Trait | Num of Hair Whorl Pigs | Mean | Num of Non-Hair Whorl Pigs | Mean | p Value |
|---|---|---|---|---|---|
| Number of Live litter size | 128 | 13.23 | 1968 | 12.21 | 0.0018 |
| Number of Total litter size | 128 | 13.94 | 1969 | 12.81 | 0.0009 |
| Number of healthy litters | 128 | 11.69 | 1968 | 10.88 | 0.0043 |
| Corrected 100 kg weight of age | 128 | 164.49 | 1723 | 159.44 | 0.0026 |
| Corrected FCR | 53 | 1.73 | 606 | 1.86 | 0.0015 |
| Birth weight | 128 | 1.22 | 1939 | 1.25 | 0.1429 |
| Number of left teats | 128 | 7.02 | 1968 | 7.05 | 0.6032 |
| Number of right teats | 128 | 6.90 | 1968 | 6.91 | 0.7724 |
| Corrected 100 kg backfat | 128 | 11.99 | 1723 | 12.16 | 0.5146 |
| Type | Hair Whorl | Non-Hair Whorl |
|---|---|---|
| Losses | 1279 (83.76%) | 17,787 (87.84%) |
| Gains | 248 (16.24%) | 2462 (12.16%) |
| Total | 1527 | 20,249 |
| SNP | SSC | Position | p Value | Alleles | Candidate Gene | Start | End |
|---|---|---|---|---|---|---|---|
| CNC10170898 | 17 | 44143739 | 1.05 × 10−6 | G/A | CHD6 | 44048098 | 44265431 |
| CNC10170895 | 17 | 44164745 | 1.32 × 10−6 | T/C | CHD6 | 44048098 | 44265431 |
| CNCB10011881 | 17 | 44126874 | 2.60 × 10−5 | C/T | CHD6 | 44048098 | 44265431 |
| CNC10023023 | 2 | 141779308 | 3.05 × 10−5 | T/G | NRG2 | 141662369 | 141824727 |
| Term | Count | p-Value | Genes |
|---|---|---|---|
| GO:2000353 positive regulation of endothelial cell apoptotic process | 2 | 0.011625061 | ECSCR, PLCG1 |
| GO:0005654 nucleoplasm | 7 | 0.022952206 | PROB1, ZHX3, STING1, SPATA24, CHD6, ECSCR, CXXC5 |
| GO:0008284 positive regulation of cell population proliferation | 3 | 0.023643003 | PURA, MZB1, HBEGF |
| GO:0098978 glutamatergic synapse | 3 | 0.031915019 | PURA, PSD2, PLCG1 |
| GO:0007173 epidermal growth factor receptor signaling pathway | 2 | 0.034496111 | PLCG1, HBEGF |
| GO:0032587 ruffle membrane | 2 | 0.063070664 | PSD2, PLCG1 |
| GO:0140297 DNA-binding transcription factor binding | 2 | 0.075270787 | PURA, CXXC5 |
| GO:0098794 postsynapse | 2 | 0.077077652 | PURA, PSD2 |
| ssc04012 ErbB signaling pathway | 3 | 0.001177119 | NRG2, PLCG1, HBEGF |
| ssc05171 Coronavirus disease—COVID-19 | 3 | 0.010096157 | STING1, PLCG1, HBEGF |
| ssc01521 EGFR tyrosine kinase inhibitor resistance | 2 | 0.04920922 | NRG2, PLCG1 |
| SNP | Number | Genotype Frequency (Hair Whorl Rate) | ||
|---|---|---|---|---|
| 0/0 | 0/1 | 1/1 | ||
| Chr17:CNC10170898 | 2625 | GG:0.8690 (0.04910) | AG:0.1265 (0.1626) | AA:0.004571 (0.4167) |
| Chr17:CNC10170895 | 2625 | TT:0.8659 (0.04927) | CT:0.1295 (0.1588) | CC:0.004571 (0.4167) |
| Chr17:CNCB10011881 | 2625 | CC:0.8270 (0.05157) | TC:0.1665 (0.1236) | TT:0.006095 (0.3125) |
| Chr2:CNC10023023 | 2625 | TT:0.8827 (0.04704) | GT:0.1070 (0.1922) | GG:0.01029 (0.2963) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, W.; Yang, X.; Zhu, L.; Yang, Y.; Liu, C.; Du, Y.; Wang, Y.; Niu, L.; Zhao, Y.; Liu, Y.; et al. Genome-Wide Association Studies of Hair Whorl in Pigs. Genes 2024, 15, 1249. https://doi.org/10.3390/genes15101249
Jiang W, Yang X, Zhu L, Yang Y, Liu C, Du Y, Wang Y, Niu L, Zhao Y, Liu Y, et al. Genome-Wide Association Studies of Hair Whorl in Pigs. Genes. 2024; 15(10):1249. https://doi.org/10.3390/genes15101249
Chicago/Turabian StyleJiang, Wenyu, Xidi Yang, Liangyu Zhu, Yiting Yang, Chengming Liu, Yong Du, Yan Wang, Lili Niu, Ye Zhao, Yihui Liu, and et al. 2024. "Genome-Wide Association Studies of Hair Whorl in Pigs" Genes 15, no. 10: 1249. https://doi.org/10.3390/genes15101249
APA StyleJiang, W., Yang, X., Zhu, L., Yang, Y., Liu, C., Du, Y., Wang, Y., Niu, L., Zhao, Y., Liu, Y., Gan, M., Shen, L., & Zhu, L. (2024). Genome-Wide Association Studies of Hair Whorl in Pigs. Genes, 15(10), 1249. https://doi.org/10.3390/genes15101249