
Diversity, Distribution and Structural Prediction of the Pathogenic Bacterial Effectors EspN and EspS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. T3SS Translocation Assay
2.2. Bioinformatics Analyses
2.3. Phylogenetic Analyses
2.4. Multiple Alignment of EspN and Its Homologs, as Well as EspS and Its Homologs
2.5. Protein Structure Prediction and Analysis
3. Results
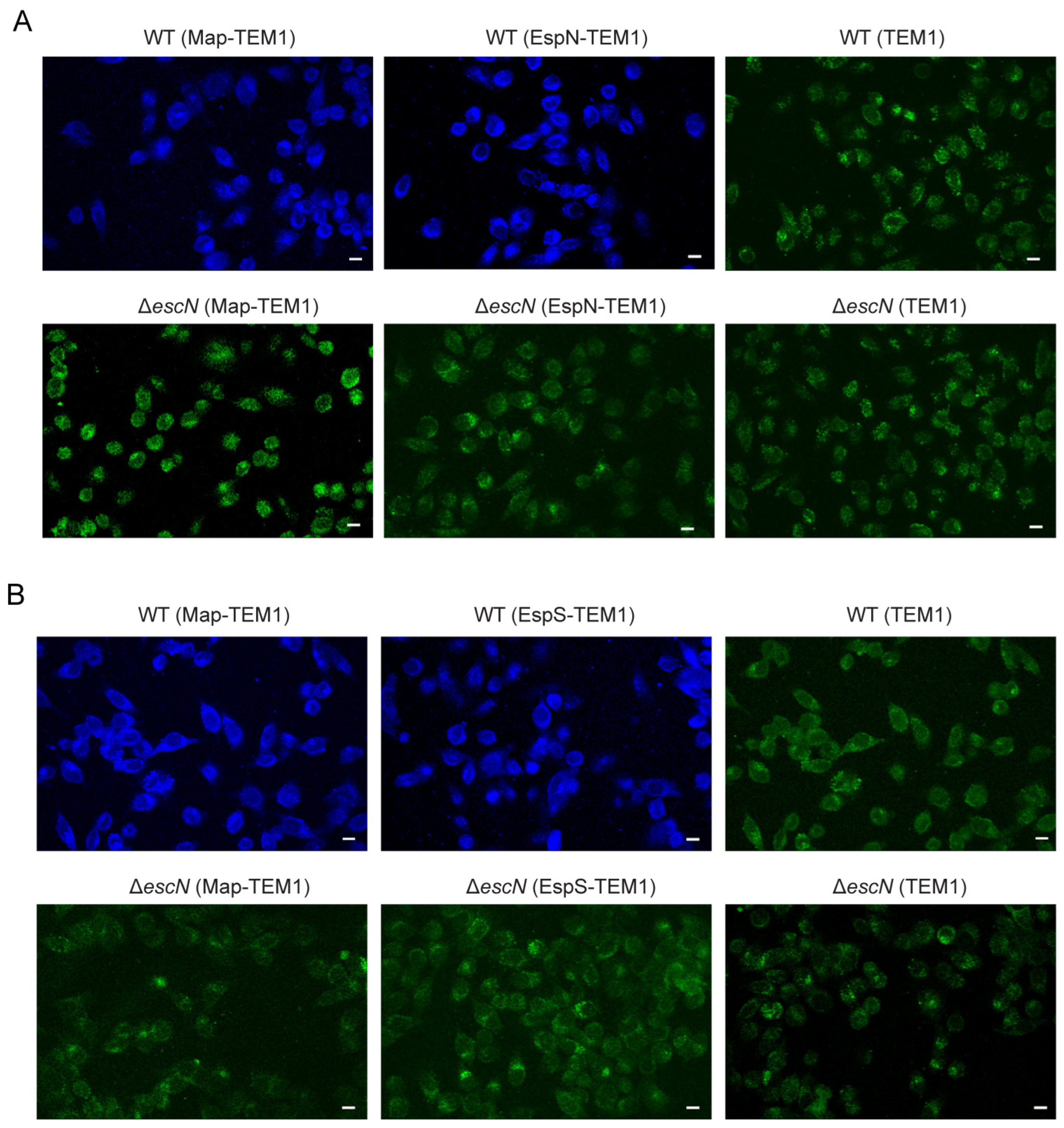
3.1. EspN or EspS Is Translocated into Host Cells by T3SS
3.2. Evolutionary Tree Analysis of EspN Revealed That Homologous Proteins of EspN Widely Distributed in Many Pathogens
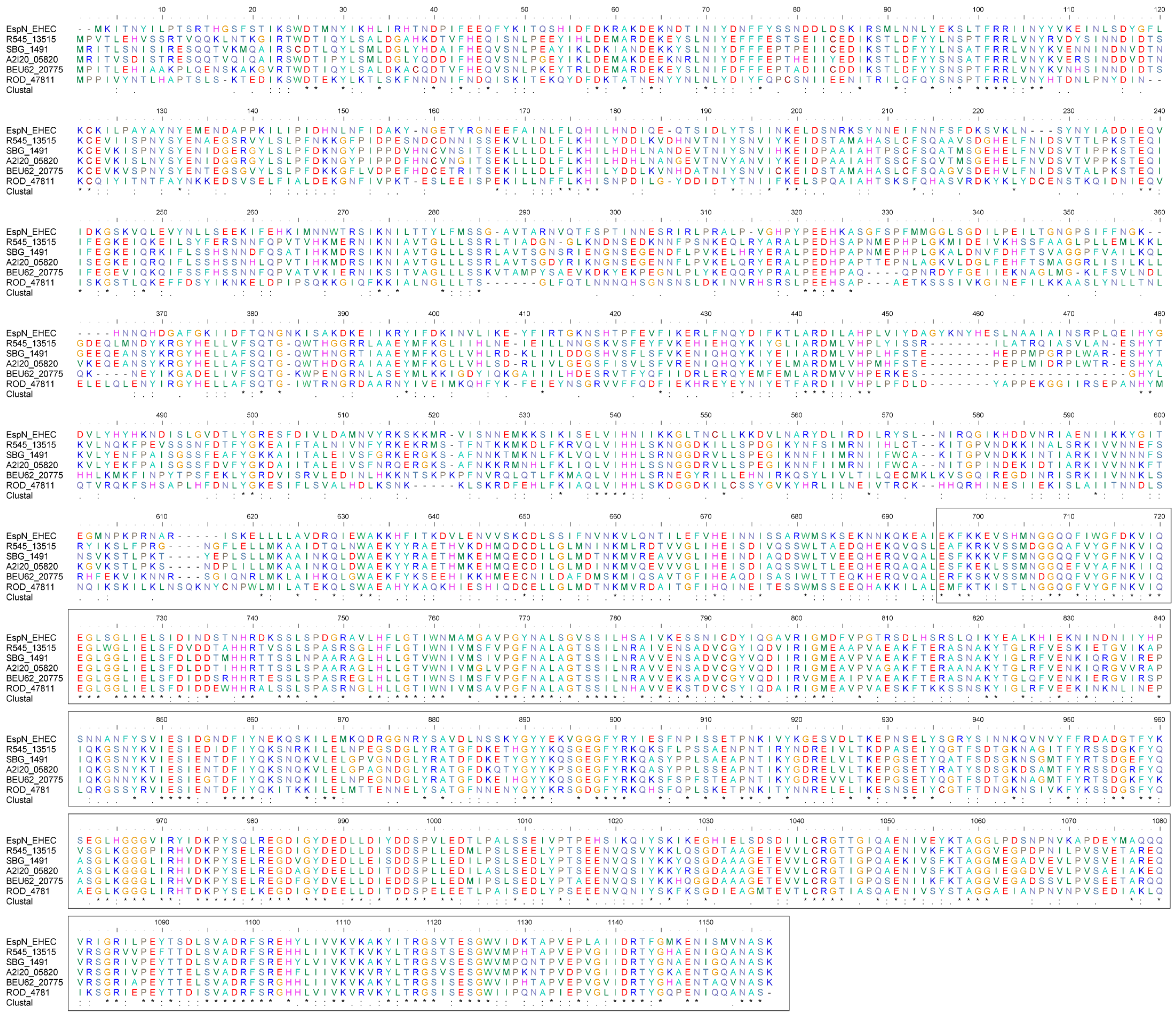
3.3. EspN and Its Homologues Share Conserved C-Terminal Sequences
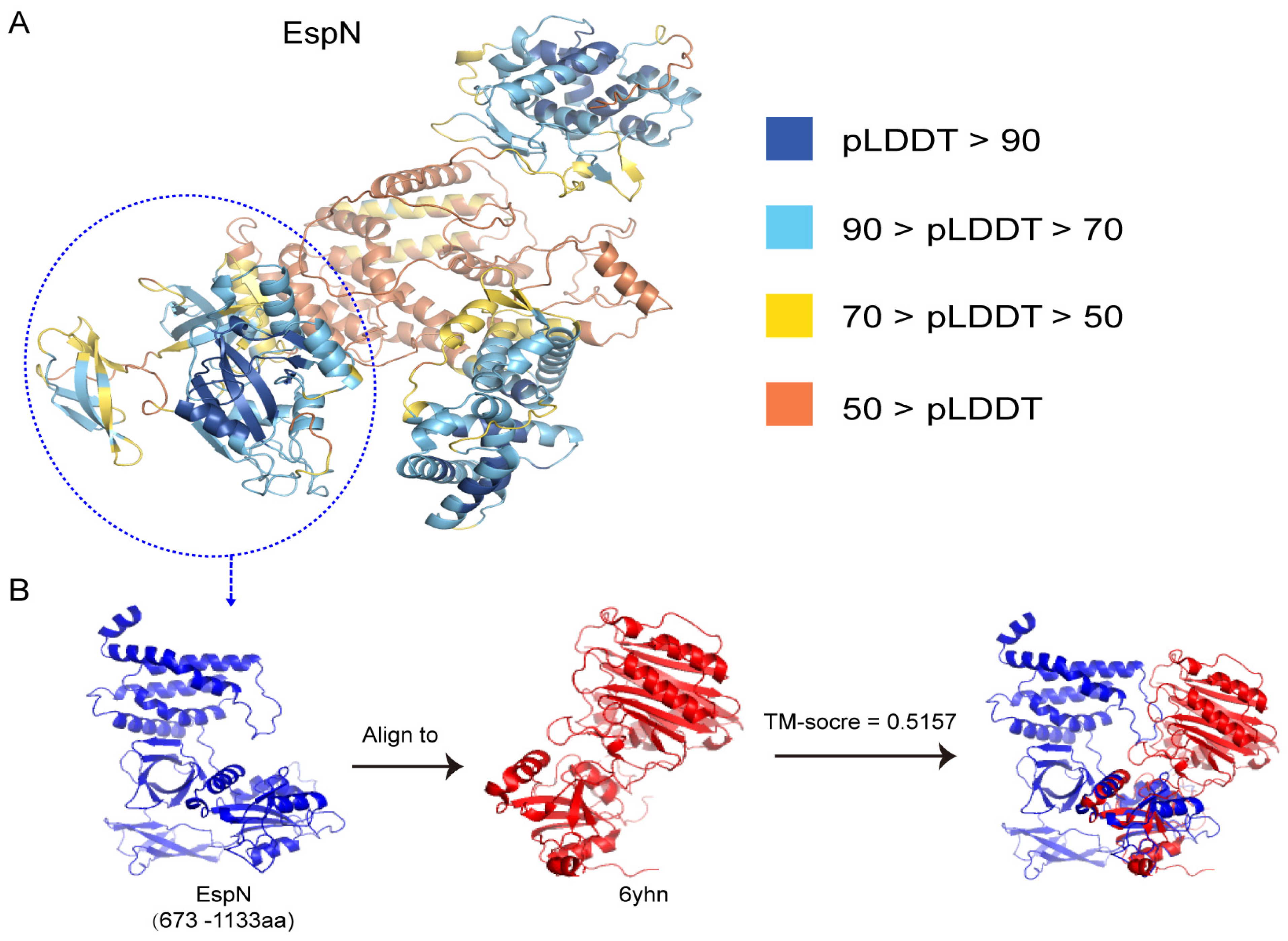
3.4. The Predicted 3D Structure of the Subsequences of EspN Shows SOME Similarity to the D4 Domain of CNF(Y)
3.5. Evolutionary Tree Analysis of EspS Revealed That Homologous Proteins of EspS Widely Distributed in Many Pathogens
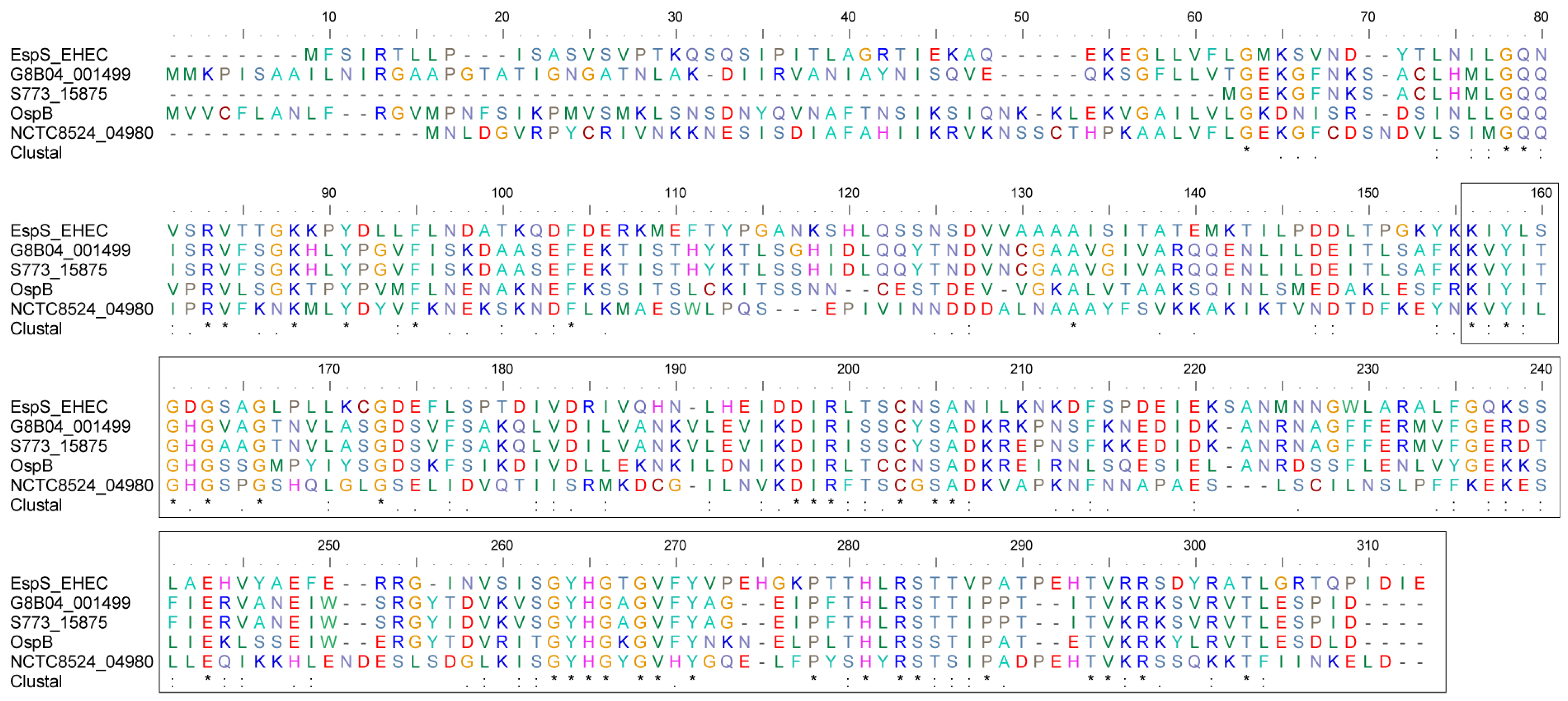
3.6. EspS and Its Homologues Share Conserved C-Terminal Sequences
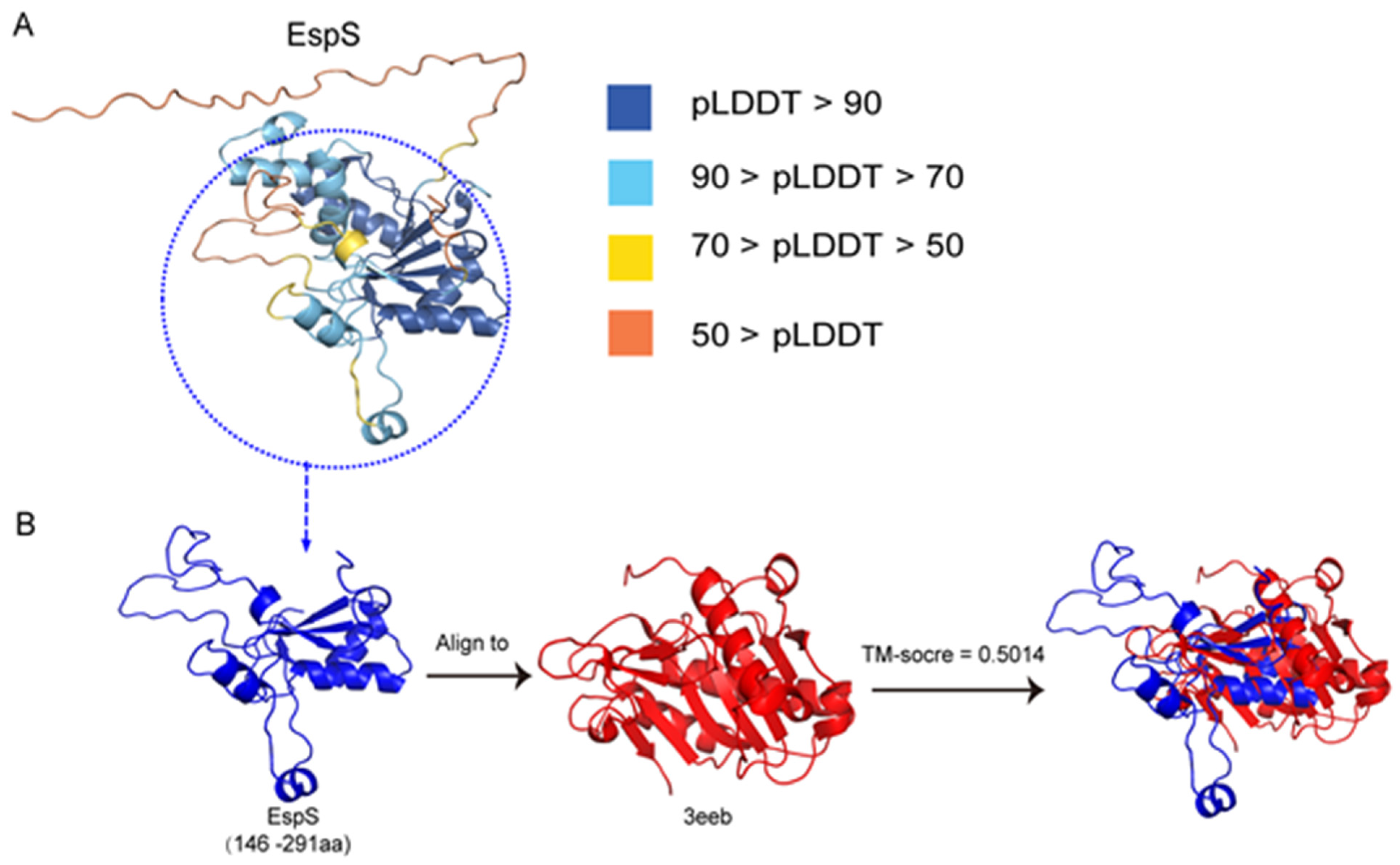
3.7. The Predicted 3D Structure of the Subsequences of EspS Shows Some Similarity to That of the Vibrio cholerae RTX Cysteine Protease Domain
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Serapio-Palacios, A.; Finlay, B.B. Dynamics of expression, secretion and translocation of type III effectors during enteropathogenic Escherichia coli infection. Curr. Opin. Microbiol. 2020, 54, 67–76. [Google Scholar] [CrossRef]
- Dean, P.; Kenny, B. The effector repertoire of enteropathogenic E. coli: Ganging up on the host cell. Curr. Opin. Microbiol. 2009, 12, 101–109. [Google Scholar] [CrossRef]
- Wong, A.R.; Pearson, J.S.; Bright, M.D.; Munera, D.; Robinson, K.S.; Lee, S.F.; Frankel, G.; Hartland, E.L. Enteropathogenic and enterohaemorrhagic Escherichia coli: Even more subversive elements. Mol. Microbiol. 2011, 80, 1420–1438. [Google Scholar] [CrossRef] [PubMed]
- Mullineaux-Sanders, C.; Sanchez-Garrido, J.; Hopkins, E.G.D.; Shenoy, A.R.; Barry, R.; Frankel, G. Citrobacter rodentium-host-microbiota interactions: Immunity, bioenergetics and metabolism. Nat. Rev. Microbiol. 2019, 17, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Dekker, J.P.; Frank, K.M. Salmonella, Shigella, and yersinia. Clin. Lab. Med. 2015, 35, 225–246. [Google Scholar] [CrossRef] [PubMed]
- Leting, S.K.; Musyoki, S.K.; Maiyoh, G.K. Characterization and drug susceptibility pattern of Salmonella and Shigella in children below five years: A cross-sectional study conducted in Lodwar, Turkana County, in Northern Kenya. Pan Afr. Med. J. 2022, 42, 13. [Google Scholar] [CrossRef]
- Pinaud, L.; Sansonetti, P.J.; Phalipon, A. Host Cell Targeting by Enteropathogenic Bacteria T3SS Effectors. Trends Microbiol. 2018, 26, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Chakravarty, A.; Guha Biswas, P.; De Guzman, R.N. The type III secretion system needle, tip, and translocon. Protein Sci. A Publ. Protein Soc. 2019, 28, 1582–1593. [Google Scholar] [CrossRef]
- Harishankar, A.; Viswanathan, V.K. Attaching and effacing pathogens modulate host mitochondrial structure and function. Int. Rev. Cell Mol. Biol. 2023, 377, 65–86. [Google Scholar] [CrossRef]
- Shenoy, A.R.; Furniss, R.C.D.; Goddard, P.J.; Clements, A. Modulation of Host Cell Processes by T3SS Effectors. Curr. Top. Microbiol. Immunol. 2018, 416, 73–115. [Google Scholar] [CrossRef]
- Cepeda-Molero, M.; Berger, C.N.; Walsham, A.D.S.; Ellis, S.J.; Wemyss-Holden, S.; Schüller, S.; Frankel, G.; Fernández, L. Attaching and effacing (A/E) lesion formation by enteropathogenic E. coli on human intestinal mucosa is dependent on non-LEE effectors. PLoS Pathog. 2017, 13, e1006706. [Google Scholar] [CrossRef] [PubMed]
- Blasche, S.; Mörtl, M.; Steuber, H.; Siszler, G.; Nisa, S.; Schwarz, F.; Lavrik, I.; Gronewold, T.M.; Maskos, K.; Donnenberg, M.S.; et al. The E. coli effector protein NleF is a caspase inhibitor. PLoS ONE 2013, 8, e58937. [Google Scholar] [CrossRef]
- Gao, X.; Wang, X.; Pham, T.H.; Feuerbacher, L.A.; Lubos, M.L.; Huang, M.; Olsen, R.; Mushegian, A.; Slawson, C.; Hardwidge, P.R. NleB, a bacterial effector with glycosyltransferase activity, targets GAPDH function to inhibit NF-κB activation. Cell Host Microbe 2013, 13, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.; Ooka, T.; Iguchi, A.; Hayashi, T.; Sugimoto, N.; Tobe, T. NleC, a type III secretion protease, compromises NF-κB activation by targeting p65/RelA. PLoS Pathog. 2010, 6, e1001231. [Google Scholar] [CrossRef] [PubMed]
- Nadler, C.; Baruch, K.; Kobi, S.; Mills, E.; Haviv, G.; Farago, M.; Alkalay, I.; Bartfeld, S.; Meyer, T.F.; Ben-Neriah, Y.; et al. The type III secretion effector NleE inhibits NF-kappaB activation. PLoS Pathog. 2010, 6, e1000743. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Mishra, P.K.K. Functional Diversity of the Excretory/Secretory Proteins of Nematode Parasites. Acta Parasitol. 2022, 67, 619–627. [Google Scholar] [CrossRef]
- Kassa, E.G.; Zlotkin-Rivkin, E.; Friedman, G.; Ramachandran, R.P.; Melamed-Book, N.; Weiss, A.M.; Belenky, M.; Reichmann, D.; Breuer, W.; Pal, R.R.; et al. Enteropathogenic Escherichia coli remodels host endosomes to promote endocytic turnover and breakdown of surface polarity. PLoS Pathog. 2019, 15, e1007851. [Google Scholar] [CrossRef]
- Ogura, Y.; Ooka, T.; Iguchi, A.; Toh, H.; Asadulghani, M.; Oshima, K.; Kodama, T.; Abe, H.; Nakayama, K.; Kurokawa, K.; et al. Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. Proc. Natl. Acad. Sci. USA 2009, 106, 17939–17944. [Google Scholar] [CrossRef]
- Petty, N.K.; Bulgin, R.; Crepin, V.F.; Cerdeño-Tárraga, A.M.; Schroeder, G.N.; Quail, M.A.; Lennard, N.; Corton, C.; Barron, A.; Clark, L.; et al. The Citrobacter rodentium genome sequence reveals convergent evolution with human pathogenic Escherichia coli. J. Bacteriol. 2010, 192, 525–538. [Google Scholar] [CrossRef]
- Li, T.; Li, Z.; Chen, F.; Liu, X.; Ning, N.; Huang, J.; Wang, H. Eukaryotic-like Kinase Expression in Enterohemorrhagic Escherichia coli: Potential for Enhancing Host Aggressive Inflammatory Response. J. Infect. Dis. 2017, 216, 1150–1158. [Google Scholar] [CrossRef]
- Allombert, J.; Vianney, A.; Charpentier, X. Monitoring Effector Translocation using the TEM-1 β-Lactamase Reporter System. Methods Mol. Biol. 2017, 1615, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Villamil, J.; Tapia-Pastrana, G.; Navarro-Garcia, F. Pathogenic Lifestyles of E. coli Pathotypes in a Standardized Epithelial Cell Model Influence Inflammatory Signaling Pathways and Cytokines Secretion. Front. Cell. Infect. Microbiol. 2016, 6, 120. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, X.; Oswald, E. Identification of the secretion and translocation domain of the enteropathogenic and enterohemorrhagic Escherichia coli effector Cif, using TEM-1 β-lactamase as a new fluorescence-based reporter. J. Bacteriol. 2004, 186, 5486–5495. [Google Scholar] [CrossRef] [PubMed]
- Mount, D.W. Using the Basic Local Alignment Search Tool (BLAST). CSH Protoc. 2007, 2007, pdb.top17. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium, T. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef]
- Baier, A.L.; Kline, A.C.; Feeny, N.C. Therapeutic alliance as a mediator of change: A systematic review and evaluation of research. Clin. Psychol. Rev. 2020, 82, 101921. [Google Scholar] [CrossRef]
- Smith, M.L.; Hahn, M.W. Phylogenetic inference using generative adversarial networks. Bioinformatics 2023, 39, btad543. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef]
- Alzohairy, A.M. BioEdit: An important software for molecular biology. Gerf Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Bittrich, S.; Wang, S.; Burley, S.K. Assessing PDB macromolecular crystal structure confidence at the individual amino acid residue level. Structure 2022, 30, 1385–1394.e1383. [Google Scholar] [CrossRef] [PubMed]
- van Kempen, M.; Kim, S.S.; Tumescheit, C.; Mirdita, M.; Lee, J.; Gilchrist, C.L.M.; Söding, J.; Steinegger, M. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 2024, 42, 243–246. [Google Scholar] [CrossRef]
- Porter, K.A.; Padhorny, D.; Desta, I.; Ignatov, M.; Beglov, D.; Kotelnikov, S.; Sun, Z.; Alekseenko, A.; Anishchenko, I.; Cong, Q.; et al. Template-based modeling by ClusPro in CASP13 and the potential for using co-evolutionary information in docking. Proteins 2019, 87, 1241–1248. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Chaoprasid, P.; Lukat, P.; Mühlen, S.; Heidler, T.; Gazdag, E.M.; Dong, S.; Bi, W.; Rüter, C.; Kirchenwitz, M.; Steffen, A.; et al. Crystal structure of bacterial cytotoxic necrotizing factor CNF(Y) reveals molecular building blocks for intoxication. EMBO J. 2021, 40, e105202. [Google Scholar] [CrossRef]
- Lupardus, P.J.; Shen, A.; Bogyo, M.; Garcia, K.C. Small molecule-induced allosteric activation of the Vibrio cholerae RTX cysteine protease domain. Science 2008, 322, 265–268. [Google Scholar] [CrossRef]
- Kenny, B.; Jepson, M. Targeting of an enteropathogenic Escherichia coli (EPEC) effector protein to host mitochondria. Cell. Microbiol. 2000, 2, 579–590. [Google Scholar] [CrossRef]
- Lemichez, E.; Aktories, K. Hijacking of Rho GTPases during bacterial infection. Exp. Cell Res. 2013, 319, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Munro, P.; Lemichez, E. Bacterial toxins activating Rho GTPases. Curr. Top. Microbiol. Immunol. 2005, 291, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Aleksiev, T.; Potestio, R.; Pontiggia, F.; Cozzini, S.; Micheletti, C. PiSQRD: A web server for decomposing proteins into quasi-rigid dynamical domains. Bioinformatics 2009, 25, 2743–2744. [Google Scholar] [CrossRef]
- Sanchez-Garrido, J.; Ruano-Gallego, D.; Choudhary, J.S.; Frankel, G. The type III secretion system effector network hypothesis. Trends Microbiol. 2022, 30, 524–533. [Google Scholar] [CrossRef]
- Deane, J.E.; Abrusci, P.; Johnson, S.; Lea, S.M. Timing is everything: The regulation of type III secretion. Cell. Mol. Life Sci. 2010, 67, 1065–1075. [Google Scholar] [CrossRef]
- Agard, D.A.; Bowman, G.R.; DeGrado, W.; Dokholyan, N.V.; Zhou, H.X. Solution of the protein structure prediction problem at last: Crucial innovations and next frontiers. Fac. Rev. 2022, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Applying and improving AlphaFold at CASP14. Proteins 2021, 89, 1711–1721. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- David, A.; Islam, S.; Tankhilevich, E.; Sternberg, M.J.E. The AlphaFold Database of Protein Structures: A Biologist’s Guide. J. Mol. Biol. 2022, 434, 167336. [Google Scholar] [CrossRef]
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef]
- Hetmann, M.; Parigger, L.; Sirelkhatim, H.; Stern, A.; Krassnigg, A.; Gruber, K.; Steinkellner, G.; Ruau, D.; Gruber, C.C. Folding the human proteome using BioNeMo: A fused dataset of structural models for machine learning purposes. Sci. Data 2024, 11, 591. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Hu, Y.; Song, Y.; Li, D.; Yang, X.; Zhang, L.; Li, T.; Wang, H. Diversity, Distribution and Structural Prediction of the Pathogenic Bacterial Effectors EspN and EspS. Genes 2024, 15, 1250. https://doi.org/10.3390/genes15101250
Li Z, Hu Y, Song Y, Li D, Yang X, Zhang L, Li T, Wang H. Diversity, Distribution and Structural Prediction of the Pathogenic Bacterial Effectors EspN and EspS. Genes. 2024; 15(10):1250. https://doi.org/10.3390/genes15101250
Chicago/Turabian StyleLi, Zhan, Yuru Hu, Yuan Song, Deyu Li, Xiaolan Yang, Liangyan Zhang, Tao Li, and Hui Wang. 2024. "Diversity, Distribution and Structural Prediction of the Pathogenic Bacterial Effectors EspN and EspS" Genes 15, no. 10: 1250. https://doi.org/10.3390/genes15101250
APA StyleLi, Z., Hu, Y., Song, Y., Li, D., Yang, X., Zhang, L., Li, T., & Wang, H. (2024). Diversity, Distribution and Structural Prediction of the Pathogenic Bacterial Effectors EspN and EspS. Genes, 15(10), 1250. https://doi.org/10.3390/genes15101250