
Multi-Omics Revealed Regulatory Mechanisms Underlying the Flowering of Ferula sinkiangensis across Three Dimensions

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. RNA Extraction and Transcriptome Sequencing
2.3. Metabolite Extraction
2.4. LC–MS/MS Analysis and Data Processing
2.5. Proteome Sequencing Analysis
2.6. Proteome-Associated LC-MS/MS Detection and Analysis
2.7. Bioinformatic Analysis
3. Results
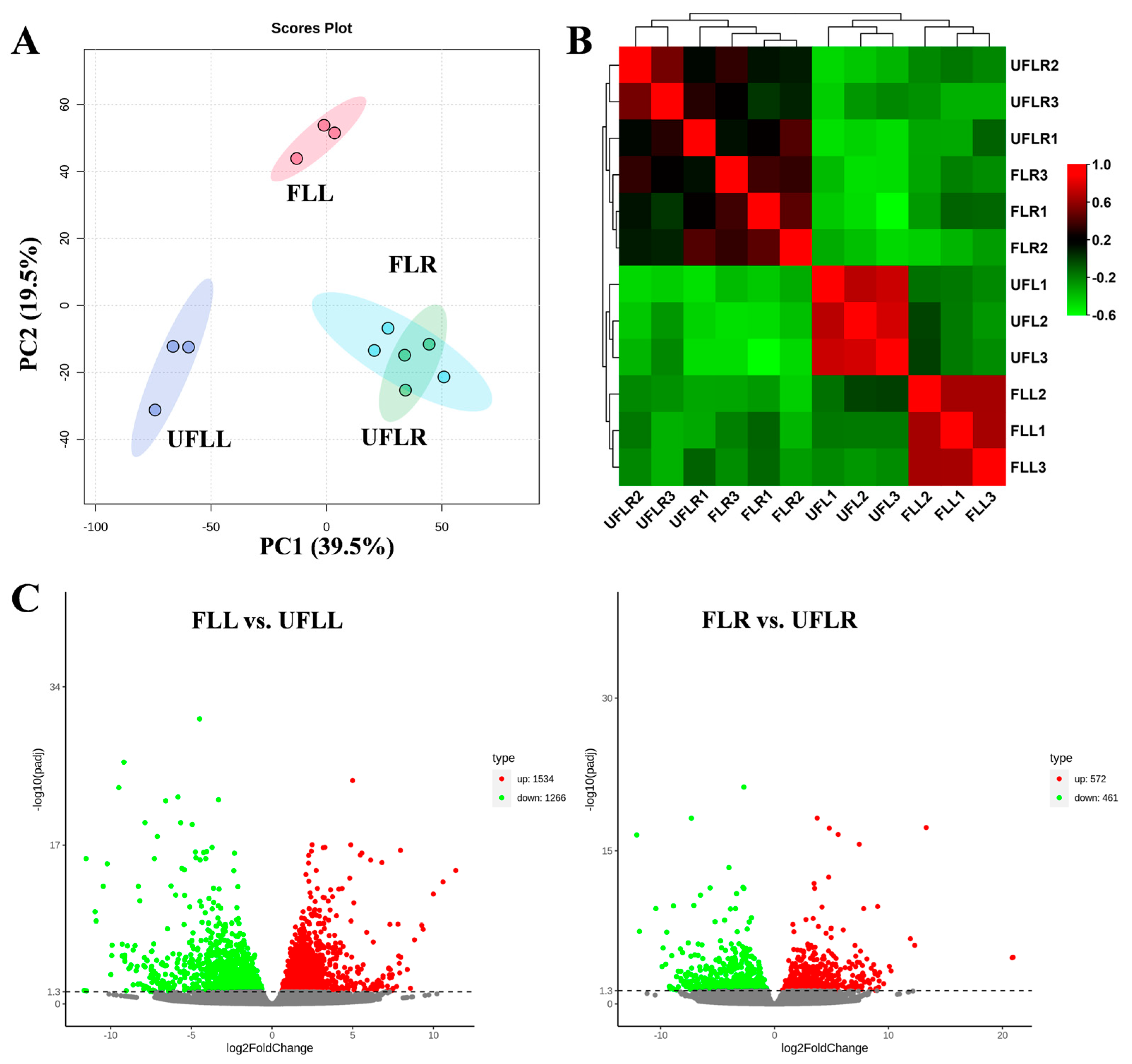
3.1. Transcriptome Variations in Leaves and Roots Associated with the Flowering Phenotype
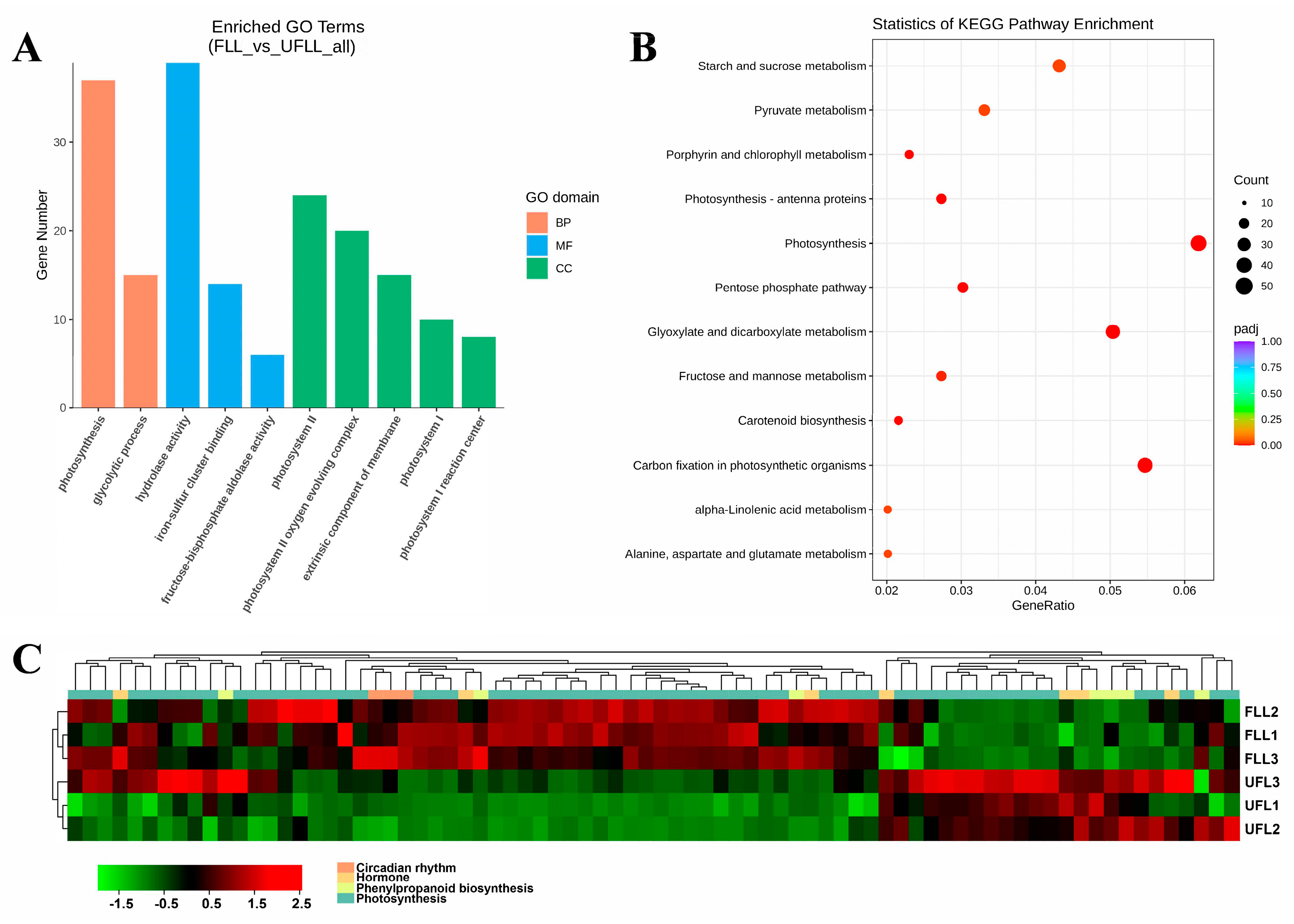
3.2. Transcriptional Changes in Phenylpropanoid, Hormones, and Photosynthesis Associated with the Flowering Phenotype
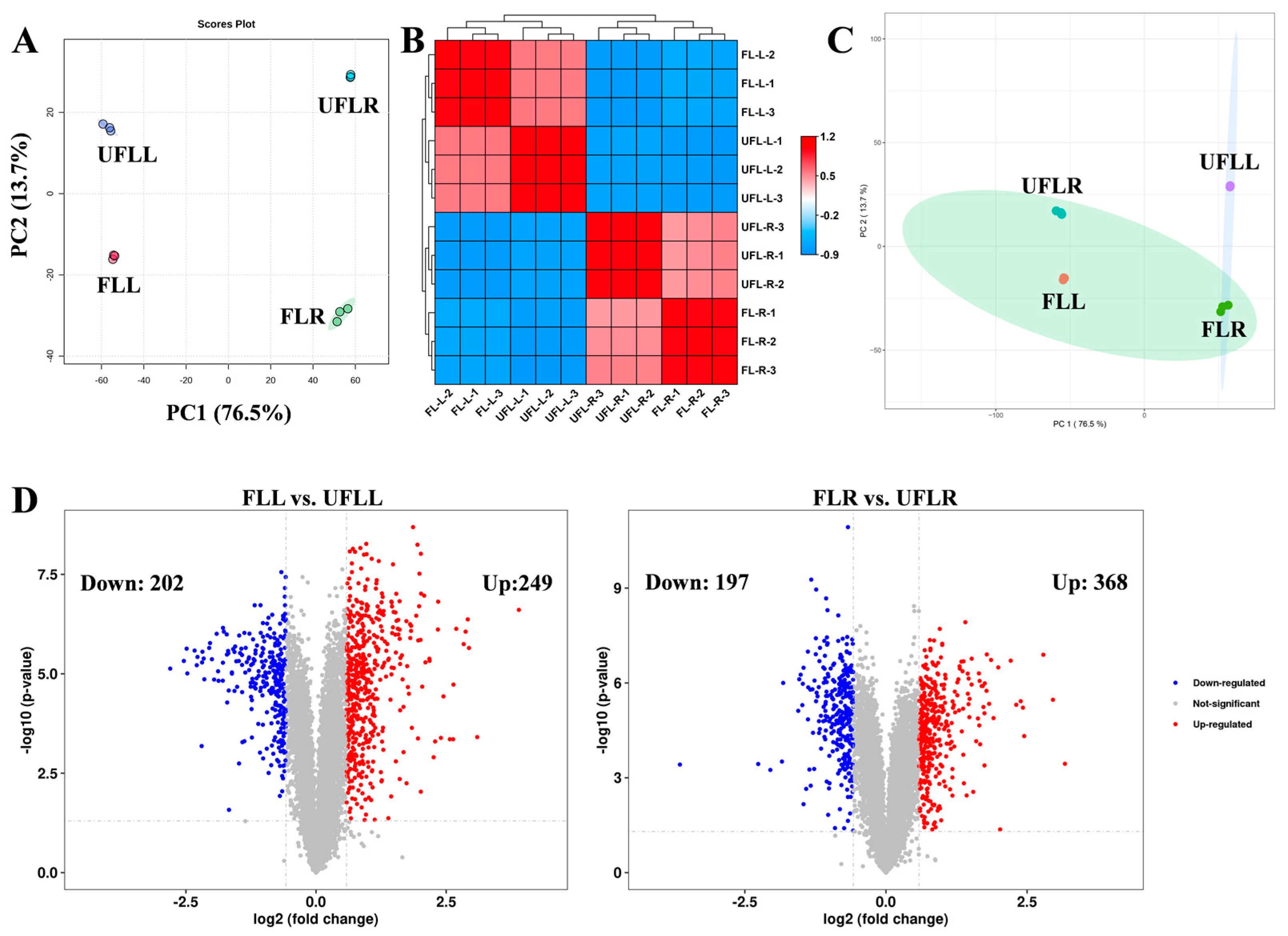
3.3. Proteome Profiling in Leaf and Root Associated with Flowering Phenotype
3.4. Proteomic Changes in Photosynthesis and Phenylpropanoid Biosynthesis in Leaves Associated with the Flowering Process of F. sinkiangensis
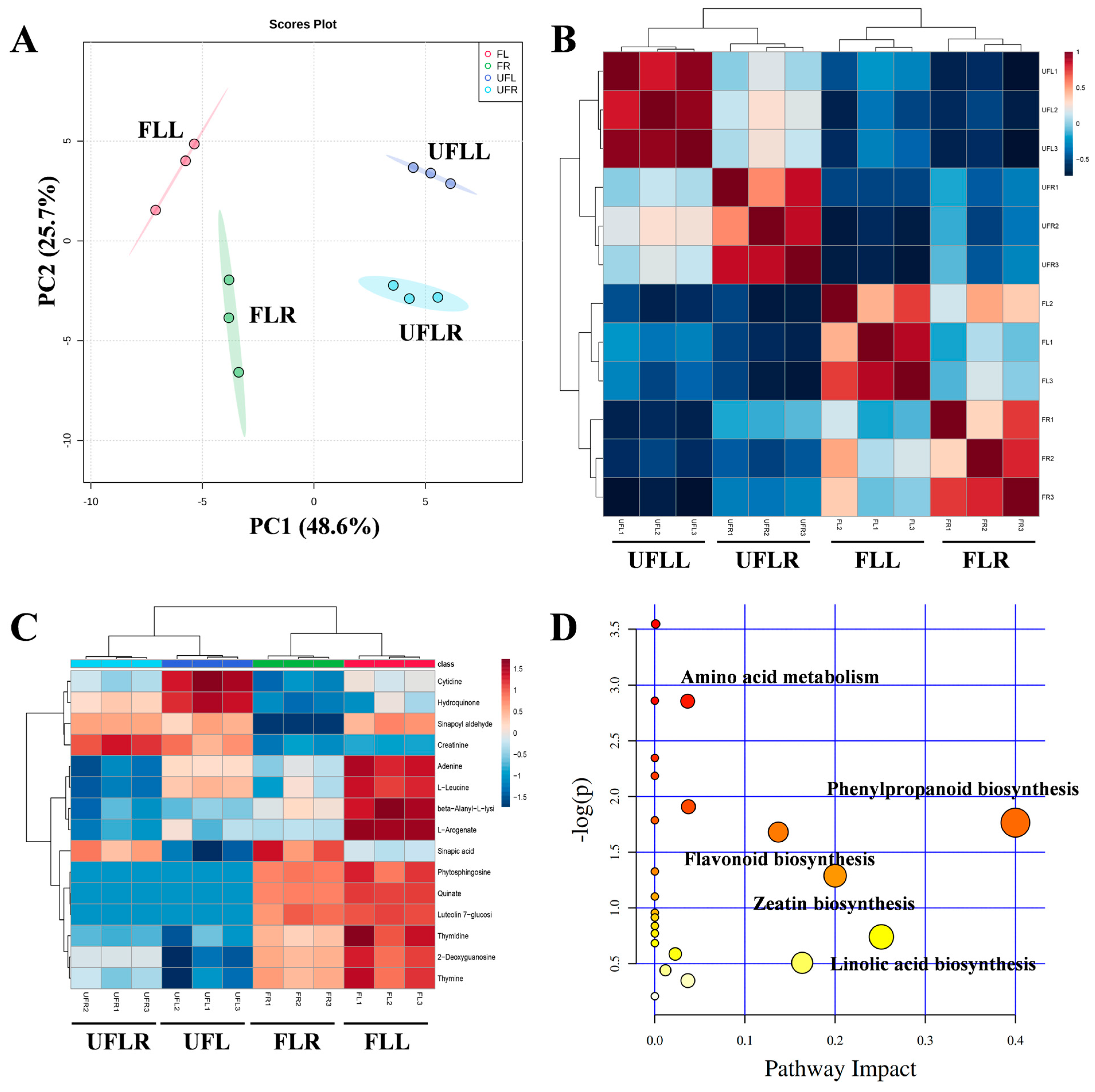
3.5. Metabolome Profiling Unveils That Phenylpropanoid Accumulation Is Associated with the Flowering Process
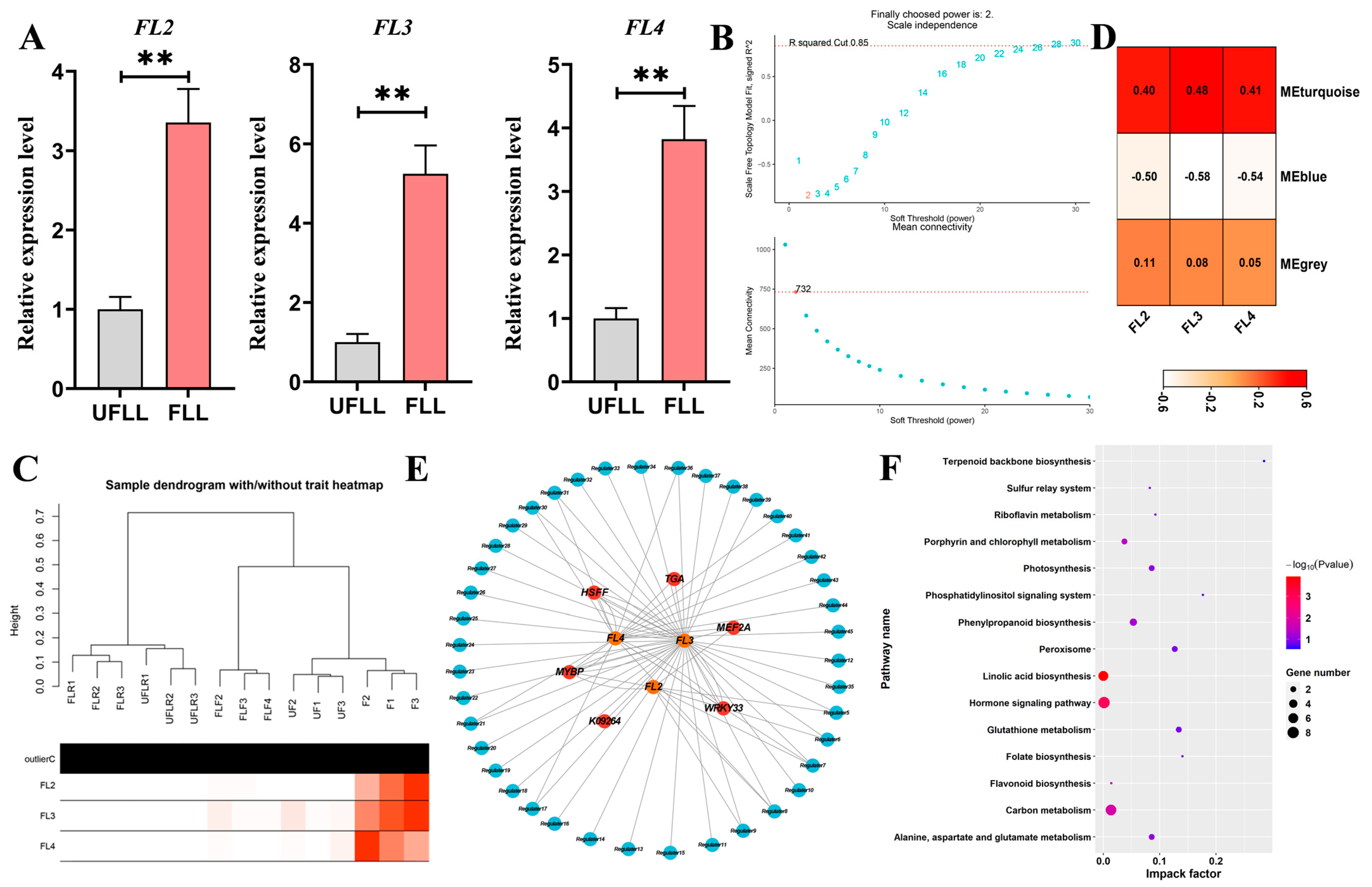
3.6. Three Genes Promote Flowering in F. sinkiangensis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lai, X.; Yang, X. Research progress of endangered medicinal plant Ferula sinkiangensis. Mod. Agric. Sci. Technol. 2022, 11, 43–47+51. [Google Scholar]
- Saeidy, S.; Nasirpour, A.; Djelveh, G.; Ursu, A.V.; Marcati, A.; Gardarin, C.; Laroche, C.; Delattre, C.; Pierre, G.; Keramat, J.; et al. Rheological and functional properties of asafoetida gum. Int. J. Biol. Macromol. 2018, 118, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
- Khayat, M.T.; Alharbi, M.; Ghazawi, K.F.; Mohamed, G.A.; Ibrahim, S.R.M. Ferula sinkiangensis (chou–AWei, Chinese Ferula): Traditional uses, phytoconstituents, biosynthesis, and pharmacological activities. Plants 2023, 4, 902. [Google Scholar] [CrossRef]
- Iranshahy, M.; Iranshahi, M. Traditional uses, phytochemistry and pharmacology of asafoetida (Ferula assa-foetida oleo-gum-resin)—A review. J. Ethnopharmacol. 2011, 134, 1–10. [Google Scholar] [CrossRef]
- Srikanth, A.; Schmid, M. Regulation of flowering time: All roads lead to Rome. Cell Mol. Life Sci. 2011, 68, 2013–2037. [Google Scholar] [CrossRef]
- Hildebrandt, T.M.; Nunes, N.A.; Araújo, W.L.; Braun, H.P. Amino acid catabolism in plants. Mol. Plant 2015, 11, 1563–1579. [Google Scholar] [CrossRef]
- Yoneyama, T.; Suzuki, A. Light–independent nitrogen assimilation in plant leaves: Nitrate incorporation into Glutamine, Glutamate, Aspartate, and Asparagine traced by 15N. Plants 2020, 10, 1303. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Zhang, Y.; Xu, G. How does nitrogen shape plant architecture? J. Exp. Bot. 2020, 15, 4415–4427. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y. Auxin biosynthesis and its role in plant development. Annu. Rev. Plant Biol. 2010, 61, 49–64. [Google Scholar] [CrossRef]
- Karami, O.; Rahimi, A. The end of flowering: Interactions between cytokinin and regulatory genes. Trends Plant Sci. 2022, 9, 840–842. [Google Scholar] [CrossRef]
- Wickland, D.P.; Hanzawa, Y. The Flowering locus T/Terminal flower 1 gene family: Functional evolution and molecular mechanisms. Mol. Plant 2015, 7, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Bai, J.; Xie, Z.; Lian, Z.; Guo, J.; Zhao, F.; Liang, Y.; Huo, H.; Gong, H. Tomato sucrose transporter SlSUT4 participates in flowering regulation by modulating gibberellin biosynthesis. Plant Physiol. 2023, 2, 1080–1098. [Google Scholar] [CrossRef] [PubMed]
- Bolouri Moghaddam, M.R.; Van den Ende, W. Sugars, the clock and transition to flowering. Front. Plant Sci. 2013, 4, 22. [Google Scholar]
- Du, Y.; Jia, H.; Yang, Z.; Wang, S.; Liu, Y.; Ma, H.; Liang, X.; Wang, B.; Zhu, M.; Meng, Y.; et al. Sufficient coumarin accumulation improves apple resistance to Cytospora mali under high–potassium status. Plant Physiol. 2023, 2, 1396–1419. [Google Scholar] [CrossRef] [PubMed]
- Peer, W.A.; Murphy, A.S. Flavonoids and auxin transport: Modulators or regulators? Trends Plant Sci. 2007, 12, 556–563. [Google Scholar] [CrossRef]
- Koshio, A.; Hasegawa, T.; Okada, R.; Takeno, K. Endogenous factors regulating poor–nutrition stress–induced flowering in pharbitis: The involvement of metabolic pathways regulated by aminooxyacetic acid. J. Plant Physiol. 2015, 173, 82–88. [Google Scholar] [CrossRef]
- Xu, N.; Wu, Z.; Li, X.; Yang, M.; Han, J.; Lu, B.; Lu, B.; Wang, J. Effects of nicosulfuron on plant growth and sugar metabolism in sweet maize (Zea mays L.). PLoS ONE 2022, 17, e0276606. [Google Scholar] [CrossRef]
- Jin, J.; Zhang, H.; Zhang, J.; Liu, P.; Chen, X.; Li, Z.; Xu, Y.; Lu, P.; Cao, P. Integrated transcriptomics and metabolomics analysis to characterize cold stress responses in Nicotiana tabacum. BMC Genom. 2017, 1, 496. [Google Scholar] [CrossRef]
- Wang, J.L.; Zhang, T.; Shen, X.T.; Liu, J.; Zhao, D.L.; Sun, Y.W.; Wang, L.; Liu, Y.J.; Gong, X.Y.; Liu, Y.X. Serum metabolomics for early diagnosis of esophageal squamous cell carcinoma by UHPLC–QTOF/MS. Metabolomics 2016, 12, 116. [Google Scholar] [CrossRef]
- Xia, J.; Sinelnikov, I.V.; Han, B.; Wishart, D.S. MetaboAnalyst 3.0--making metabolomics more meaningful. Nucleic Acids Res. 2015, 43, W251–W257. [Google Scholar] [CrossRef]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 9, P3. [Google Scholar]
- Plubell, D.L.; Wilmarth, P.A.; Zhao, Y.; Fenton, A.M.; Minnier, J.; Reddy, A.P.; Klimek, J.; Yang, X.; David, L.L.; Pamir, N. Extended multiplexing of Tandem Mass Tags (TMT) labeling reveals age and high fat diet specific proteome changes in mouse epididymal adipose tissue. Mol. Cell Proteom. 2017, 5, 873–890. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, J.; Gao, L.; Liu, H.; Liu, C. iTRAQ–Based proteomic analysis of neonatal kidney from offspring of protein restricted rats reveals abnormalities in intraflagellar transport proteins. Cell Physiol. Biochem. 2017, 1, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Walter, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 27, 29–34. [Google Scholar] [CrossRef]
- Du, Y.; Liu, G.; Jia, H.; Liu, Y.; Tan, Y.; Wang, S.; Mu, J.; Yu, J.; Xue, K.; Zhang, R.; et al. Changes in planta K nutrient content altered the interaction pattern between Nicotiana benthamiana and Alternaria longipes. Plant Cell Environ. 2024, 9, 3619–3637. [Google Scholar] [CrossRef]
- Sibout, R.; Plantegenet, S.; Hardtke, C.S. Flowering as a condition for xylem expansion in Arabidopsis hypocotyl and root. Curr. Biol. 2008, 6, 458–463. [Google Scholar] [CrossRef]
- Xu, B.; Sathitsuksanoh, N.; Tang, Y.H.; Udvardi, M.K.; Zhang, J.Y.; Shen, Z.X.; Balota, M.; Harich, K.; Zhang, P.Y.; Zhao, B.Y. Overexpression of AtLOV1 in Switchgrass alters plant architecture, lignin content, and flowering time. PLoS ONE 2012, 12, e47399. [Google Scholar] [CrossRef]
- Cho, L.H.; Pasriga, R.; Yoon, J.; Jeon, J.S.; An, G. Roles of sugars in controlling flowering time. J. Plant Biol. 2018, 3, 121–130. [Google Scholar] [CrossRef]
- Quiroz, S.; Yustis, J.C.; Chávez–Hernández, E.C.; Martínez, T.; de la Paz, S.M.; Garay–Arroyo, A.; Álvarez–Buylla, E.R.; García–Ponce, B. Beyond the genetic pathways, flowering regulation complexity in Arabidopsis thaliana. Int. J. Mol. Sci. 2021, 11, 5716. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Hackel, A.; Kühn, C. Expression level of mature miR172 in wild type and StSUT4–silenced plants of Solanum tuberosum is sucrose–dependent. Int. J. Mol. Sci. 2021, 3, 1455. [Google Scholar] [CrossRef] [PubMed]
- Haydon, M.J.; Mielczarek, O.; Frank, A.; Roman, A.; Webb, A.A.R. Sucrose and ethylene signaling interact to modulate the circadian clock. Plant Physiol. 2017, 2, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Nohales, M.A.; Kay, S.A. GIGANTEA gates gibberellin signaling through stabilization of the DELLA proteins in Arabidopsis. Proc. Natl. Acad. Sci. USA 2019, 43, 21893–21899. [Google Scholar] [CrossRef] [PubMed]
- Fornara, F.; de Montaigu, A.; Coupland, G. SnapShot: Control of flowering in Arabidopsis. Cell 2010, 3, 550. [Google Scholar] [CrossRef]
- Matías-Hernández, L.; Aguilar-Jaramillo, A.E.; Cigliano, R.A.; Sanseverino, W.; Pelaz, S. Flowering and trichome development share hormonal and transcription factor regulation. J. Exp. Bot. 2016, 67, 1209–1219. [Google Scholar] [CrossRef]
- Ma, Z.B.; Li, W.; Wang, H.P.; Yu, D.Q. WRKY transcription factors WRKY12 and WRKY13 interact with SPL10 to modulate age–mediated flowering. J. Integr. Plant Biol. 2020, 11, 1659–1673. [Google Scholar] [CrossRef]
- Huang, R.; Sui, S.Z.; Liu, H.M.; Li, M.Y.; Liu, D.F. Overexpression of CpWRKY75 from Chimonanthus praecox promotes flowering time in transgenic Arabidopsis. Genes 2021, 1, 68. [Google Scholar] [CrossRef]
- Wang, J.C.; Wu, F.Q.; Zhu, S.S.; Xu, Y.; Cheng, Z.J.; Wang, J.L.; Li, C.N.; Sheng, P.K.; Zhang, H.; Cai, M.H.; et al. Overexpression of OsMYB1R1–VP64 fusion protein increases grain yield in rice by delaying flowering time. FEBS Lett. 2016, 19, 3385–3396. [Google Scholar] [CrossRef]
- Yi, J.X.; Derynck, M.R.; Li, X.Y.; Telmer, P.; Marsolais, F.; Dhaubhadel, S. A single–repeat MYB transcription factor, GmMYB176, regulates CHS8 gene expression and affects isoflavonoid biosynthesis in soybean. Plant J. 2010, 6, 1019–1034. [Google Scholar] [CrossRef] [PubMed]
- Amato, A.; Cavallini, E.; Zenoni, S.; Finezzo, L.; Begheldo, M.; Ruperti, B.; Tornielli, G.B. A Grapevine TTG2-like WRKY transcription factor is involved in regulating vacuolar transport and flavonoid biosynthesis. Front. Plant Sci. 2017, 7, 1979. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Jia, R.; Jiang, Y.; Fu, J.; Dong, T.; Deng, J.; Zhang, L. Functional verification of the JmLFY gene associated with the flowering of Juglans mandshurica Maxim. PeerJ 2023, 11, e14938. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Li, S.; Deng, S.; Wang, M.; Wu, Y.; Li, M.; Dong, J.; Lu, S.; Su, C.; Li, G.; et al. A method of genetic transformation and gene editing of succulents without tissue culture. Plant Biotechnol. J. 2024, 7, 1981–1988. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, C.; Li, Y.; Zhang, J.; Zhao, Y.; Zhang, Y.; Zhu, J.; Gao, X.; Liang, Y.; Qiu, Y.; Song, J.; et al. Multi-Omics Revealed Regulatory Mechanisms Underlying the Flowering of Ferula sinkiangensis across Three Dimensions. Genes 2024, 15, 1275. https://doi.org/10.3390/genes15101275
Fan C, Li Y, Zhang J, Zhao Y, Zhang Y, Zhu J, Gao X, Liang Y, Qiu Y, Song J, et al. Multi-Omics Revealed Regulatory Mechanisms Underlying the Flowering of Ferula sinkiangensis across Three Dimensions. Genes. 2024; 15(10):1275. https://doi.org/10.3390/genes15101275
Chicago/Turabian StyleFan, Congzhao, Yanfei Li, Jizhao Zhang, Yaqin Zhao, Yigong Zhang, Jun Zhu, Xingwang Gao, Yan Liang, Yuanjin Qiu, Jingyuan Song, and et al. 2024. "Multi-Omics Revealed Regulatory Mechanisms Underlying the Flowering of Ferula sinkiangensis across Three Dimensions" Genes 15, no. 10: 1275. https://doi.org/10.3390/genes15101275