
DNA Methylation Participates in Drought Stress Memory and Response to Drought in Medicago ruthenica

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
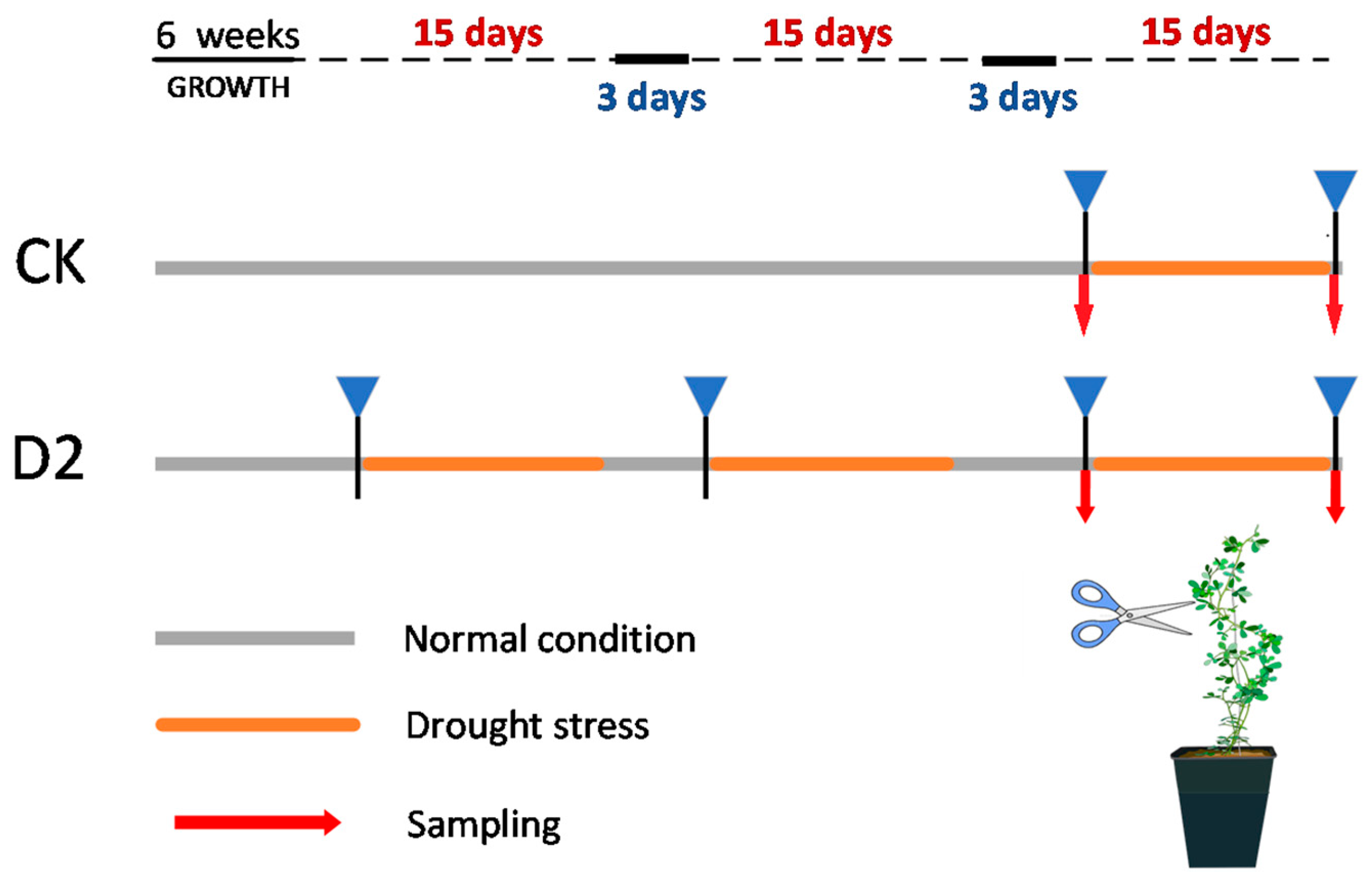
2.1. Plant Material and Drought Treatments
2.2. Morphological Traits
2.3. Leaf Physiological Variable Measurement
2.4. Soluble Sugar Content
2.5. Malondialdehyde (MDA) Content
2.6. Proline Content
2.7. ABA Content
2.8. DNA Library Construction and Whole-Genome Bisulfite Sequencing
2.9. Genome-Wide Methylation Level Analysis
2.10. RNA Extraction and Transcriptome Sequencing
2.11. Transcriptome Data Analysis
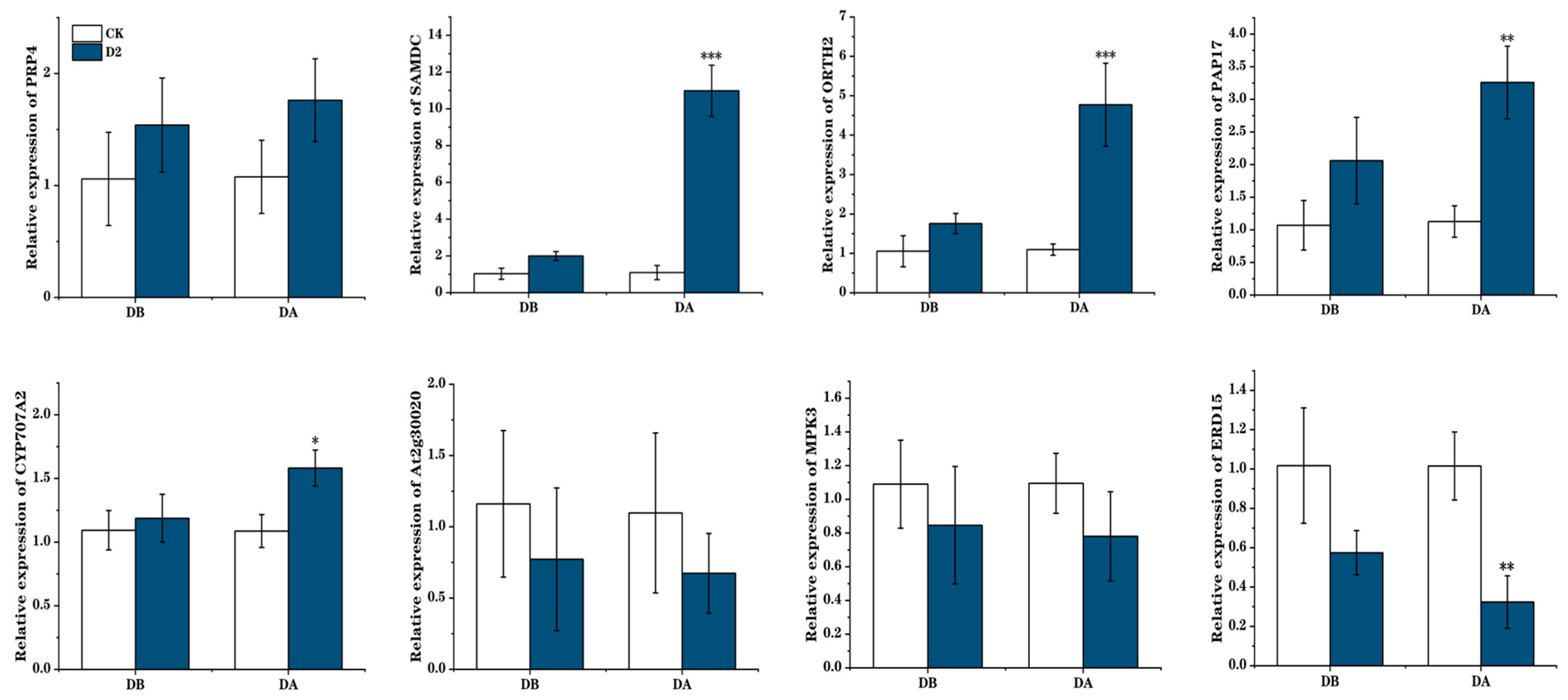
2.12. Quantitative Real-Time PCR Analysis
2.13. Statistical Analysis
3. Results
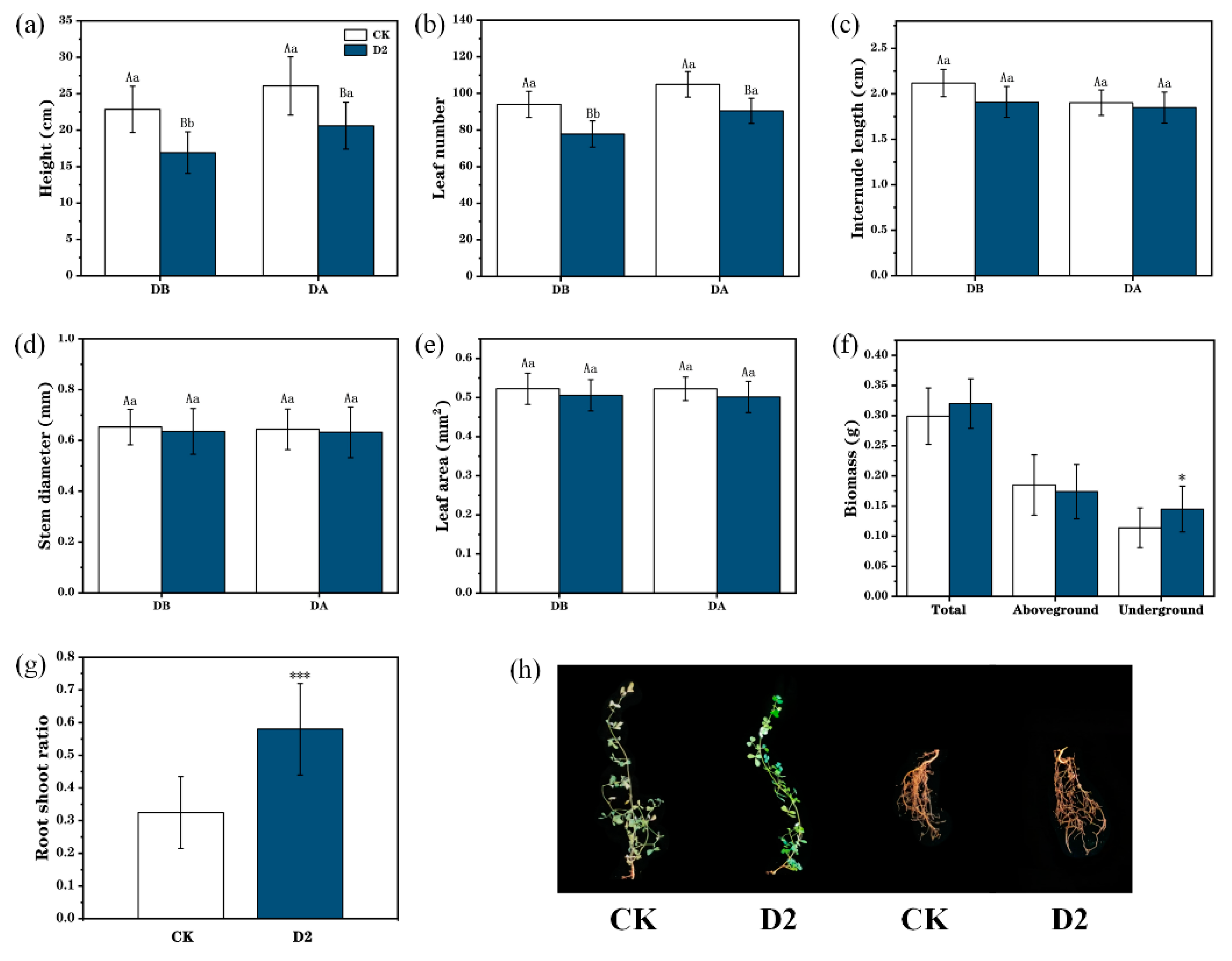
3.1. Phenotypic Changes Reveal the Response of M. ruthenica to Drought Stress
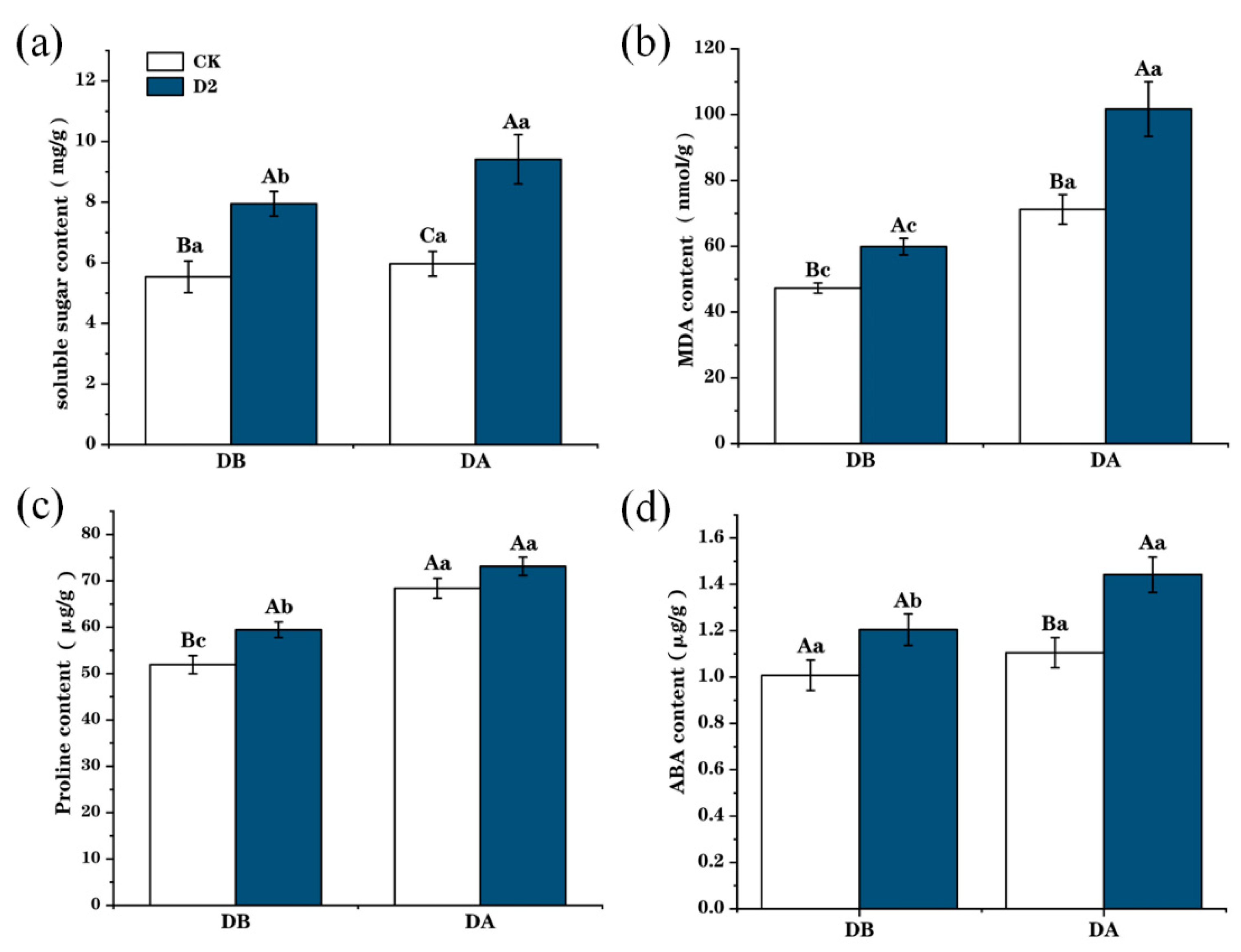
3.2. Changes in Physiological Traits of M. ruthenica in Response to Drought Stress
3.3. Changes in DEGs of M. ruthenica under Drought Stress
3.4. Methylation Landscape of M. ruthenica under Drought Stress
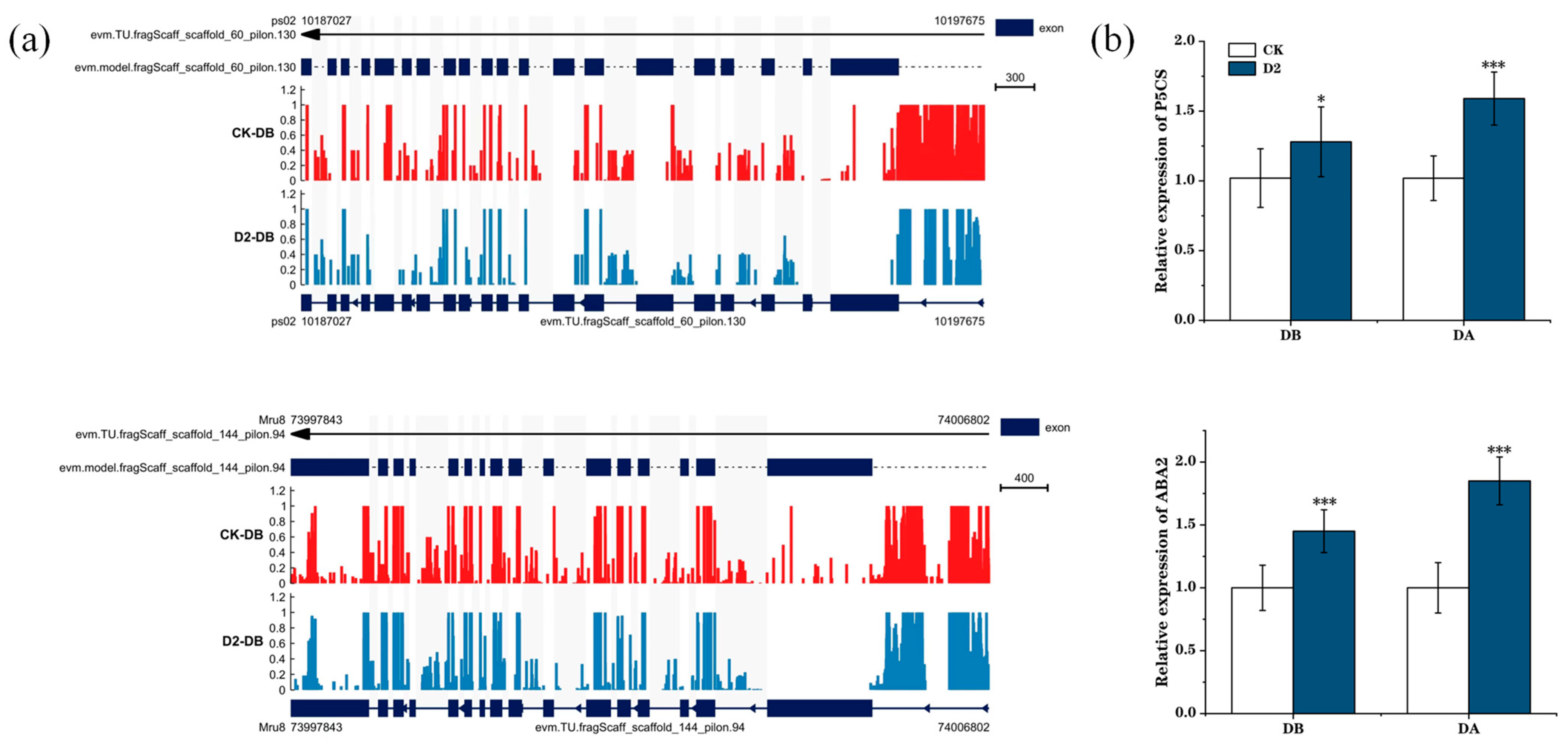
3.5. Relationship between Methylation Levels and Gene Expression under Drought Stress
4. Discussion
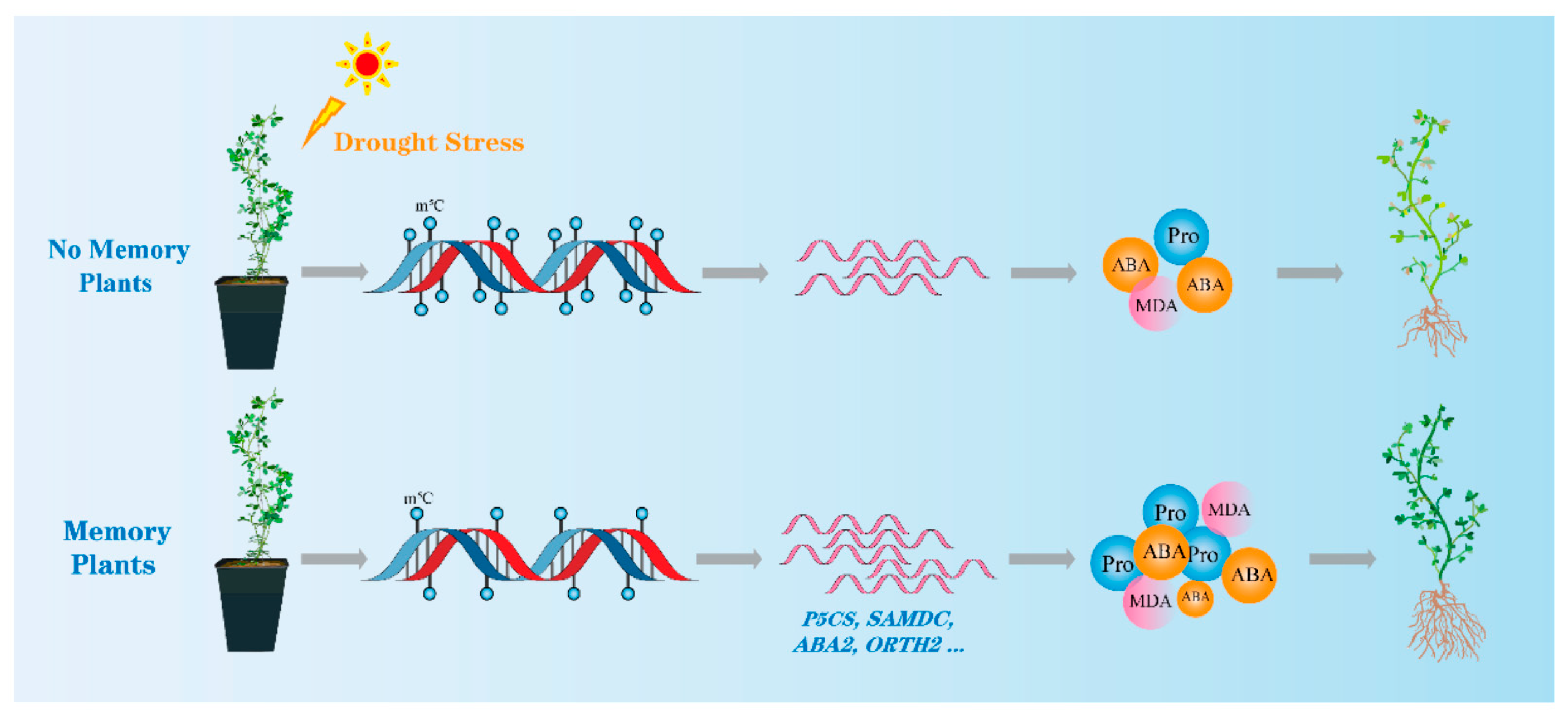
4.1. M. ruthenica Plants Actively Respond to Drought Stress, Due to Stress Memory
4.2. DNA Methylation May Contribute to Plants Stress Memory Formation
4.3. Relationship between DNA Methylation and Gene Expression
4.4. The Role of Memory Genes in Maintaining M. ruthenica Drought Stress Memory
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Touma, D.; Ashfaq, M.; Nayak, M.A.; Kao, S.C.; Diffenbaugh, N.S. A multi-model and multi-index evaluation of drought characteristics in the 21st century. J. Hydrol. 2015, 526, 196–207. [Google Scholar] [CrossRef]
- Schwalm, C.R.; Anderegg, W.R.L.; Michalak, A.M.; Fisher, J.B.; Biondi, F.; Koch, G.; Litvak, M.; Ogle, K.; Shaw, J.D.; Wolf, A.; et al. Global patterns of drought recovery. Nature 2017, 548, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Godwin, J.; Farrona, S. Plant Epigenetic Stress Memory Induced by Drought: A Physiological and Molecular Perspective. Methods Mol. Biol. 2020, 2093, 243–259. [Google Scholar] [CrossRef]
- Ciais, P.; Reichstein, M.; Viovy, N.; Granier, A.; Ogée, J.; Allard, V. Europe-wide reduction in primary productivity caused by the heat and drought in 2003. Nature 2005, 437, 529–533. [Google Scholar] [CrossRef]
- Shi, R.; Jiao, W.; Yun, L.; Zhang, Z.; Zhang, X.; Wang, Q.; Li, Y.; Mi, F. Utilization of Transcriptome, Small RNA, and Degradome Sequencing to Provide Insights into Drought Stress and Rewatering Treatment in Medicago ruthenica. Front. Plant Sci. 2021, 12, 675903. [Google Scholar] [CrossRef]
- Bruce, T.J.A.; Matthes, M.C.; Napier, J.A.; Pickett, J.A. Stressful “memories” of plants: Evidence and possible mechanisms. Plant Sci. 2007, 173, 603–608. [Google Scholar] [CrossRef]
- Walter, J.; Jentsch, A.; Beierkuhnlein, C.; Kreyling, J. Ecological stress memory and cross stress tolerance in plants in the face of climate extremes. Environ. Exp. Bot. 2013, 94, 3–8. [Google Scholar] [CrossRef]
- Oñate, M.; Blanc, J.; Munné-Bosch, S. Influence of stress history on the response of the dioecious plant Urtica dioica L. to abiotic stress. Plant Ecol. Divers. 2011, 4, 45–54. [Google Scholar] [CrossRef]
- Walter, J.; Nagy, L.; Hein, R.; Rascher, U.; Beierkuhnlein, C.; Willner, E.; Jentsch, A. Do plants remember drought? Hints towards a drought-memory in grasses. Environ. Exp. Bot. 2011, 71, 34–40. [Google Scholar] [CrossRef]
- Knight, H.; Brandt, S.; Knight, M.R. A history of stress alters drought calcium signalling pathways in Arabidopsis. Plant J. 1998, 16, 681–687. [Google Scholar] [CrossRef]
- Ding, Y.; Liu, N.; Virlouvet, L.; Riethoven, J.J.; Fromm, M.; Avramova, Z. Four distinct types of dehydration stress memory genes in Arabidopsis thaliana. BMC Plant Biol. 2013, 13, 229. [Google Scholar] [CrossRef] [PubMed]
- Beckers, G.J.M.; Conrath, U. Priming for stress resistance: From the lab to the field. Curr. Opin. Plant Biol. 2007, 10, 425–431. [Google Scholar] [CrossRef]
- Frost, C.J.; Mescher, M.C.; Carlson, J.E.; De Moraes, C.M. Plant Defense Priming against Herbivores: Getting Ready for a Different Battle. Plant Physiol. 2008, 146, 818–824. [Google Scholar] [CrossRef]
- Jaskiewicz, M.; Conrath, U.; Peterhänsel, C. Chromatin modification acts as a memory for systemic acquired resistance in the plant stress response. EMBO Rep. 2010, 12, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Goh, C.-H.; Nam, H.G.; Park, Y.S. Stress memory in plants: A negative regulation of stomatal response and transient induction of rd22 gene to light in abscisic acid-entrained Arabidopsis plants. Plant J. 2003, 36, 240–255. [Google Scholar] [CrossRef]
- Ding, Y.; Virlouvet, L.; Liu, N.; Riethoven, J.J.; Fromm, M.; Avramova, Z. Dehydration stress memory genes of Zea mays; comparison with Arabidopsis thaliana. BMC Plant Biol. 2014, 14, 141. [Google Scholar] [CrossRef] [PubMed]
- Grossniklaus, U.; Kelly, W.G.; Ferguson-Smith, A.C.; Pembrey, M.; Lindquist, S. Transgenerational epigenetic inheritance: How important is it? Nat. Rev. Genet. 2013, 14, 228. [Google Scholar] [CrossRef]
- Liu, N.; Staswick, P.E.; Avramova, Z. Memory responses of jasmonic acid-associated Arabidopsis genes to a repeated dehydration stress. Plant Cell Environ. 2016, 39, 2515–2529. [Google Scholar] [CrossRef]
- Liu, N.; Fromm, M.; Avramova, Z. H3K27me3 and H3K4me3 Chromatin Environment at Super-Induced Dehydration Stress Memory Genes of Arabidopsis thaliana. Mol. Plant. 2014, 7, 502–513. [Google Scholar] [CrossRef]
- Crisp, P.A.; Ganguly, D.; Eichten, S.R.; Borevitz, J.O.; Pogson, B.J. Reconsidering plant memory: Intersections between stress recovery, RNA turnover, and epigenetics. Sci. Adv. 2016, 2, e1501340. [Google Scholar] [CrossRef]
- Probst, A.V.; Scheid, O.M. Stress-induced structural changes in plant chromatin. Curr. Opin. Plant Biol. 2015, 27, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Hemenway, E.A.; Gehring, M. Epigenetic Regulation During Plant Development and the Capacity for Epigenetic Memory. Annu. Rev. Plant Biol. 2023, 74, 87–109. [Google Scholar] [CrossRef]
- Chang, Y.N.; Zhu, C.; Jiang, J.; Zhang, H.; Zhu, J.K.; Duan, C.G. Epigenetic regulation in plant abiotic stress responses. J. Integr. Agric. 2020, 62, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ren, L.; Li, C.; Zhang, D.; Zhang, X.; Zhou, G.; Gao, D.; Chen, R.; Chen, Y.; Wang, Z.; et al. The genome of a wild Medicago species provides insights into the tolerant mechanisms of legume forage to environmental stress. BMC Biol. 2021, 19, 96. [Google Scholar] [CrossRef] [PubMed]
- Small, E.; Jomphe, M. A synopsis of the genus Medicago leguminosae. Can. J. Bot. 1989, 67, 3260–3294. [Google Scholar] [CrossRef]
- Yin, M.; Zhang, S.; Du, X.; Mateo, R.G.; Guo, W.; Li, A.; Wang, Z.; Wu, S.; Chen, J.; Liu, J.; et al. Genomic analysis of Medicago ruthenica provides insights into its tolerance to abiotic stress and demographic history. Mol. Ecol. Resour. 2021, 21, 1641–1657. [Google Scholar] [CrossRef]
- Qu, K.; Cheng, Y.; Gao, K.; Ren, W.; Fry, E.L.; Yin, J.; Liu, Y. Growth-Defense Trade-Offs Induced by Long-term Overgrazing Could Act as a Stress Memory. Front. Plant Sci. 2022, 13, 917354. [Google Scholar] [CrossRef]
- Yilamujiang, A.; Reichelt, M.; Mithöfer, A. Slow food: Insect prey and chitin induce phytohormone accumulation and gene expression in carnivorous Nepenthes plants. Ann. Bot. 2016, 118, 369–375. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Xi, Y.; Li, W. BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genom. Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Haider, S.; Iqbal, J.; Shaukat, M.; Naseer, S.; Mahmood, T. The epigenetic chromatin-based regulation of somatic heat stress memory in plants. Plant Gene 2021, 27, 100318. [Google Scholar] [CrossRef]
- Mantoan, L.P.B.; Corrêa, C.V.; Rainho, C.A.; de Almeida, L.F.R. Rapid dehydration induces long-term water deficit memory in sorghum seedlings: Advantages and consequences. Environ. Exp. Bot. 2020, 180, 104252. [Google Scholar] [CrossRef]
- Kou, S.; Gu, Q.; Duan, L.; Liu, G.; Yuan, P.; Li, H.; Wu, Z.; Liu, W.; Huang, P.; Liu, L. Genome-Wide Bisulphite Sequencing Uncovered the Contribution of DNA Methylation to Rice Short-Term Drought Memory Formation. J. Plant Growth Regul. 2021, 41, 2903–2917. [Google Scholar] [CrossRef]
- Ventouris, Y.E.; Tani, E.; Avramidou, E.V.; Abraham, E.M.; Chorianopoulou, S.N.; Vlachostergios, D.N.; Papadopoulos, G.; Kapazoglou, A. Recurrent Water Deficit and Epigenetic Memory in Medicago sativa L. Varieties. Appl. Sci. 2020, 10, 3110. [Google Scholar] [CrossRef]
- Mirouze, M.; Paszkowski, J. Epigenetic contribution to stress adaptation in plants. Curr. Opin. Plant Biol. 2011, 14, 267–274. [Google Scholar] [CrossRef]
- Bräutigam, K.; Vining, K.J.; Lafon-Placette, C.; Fossdal, C.G.; Mirouze, M.; Marcos, J.G.; Fluch, S.; Fraga, M.F.; Guevara, M.Á.; Abarca, D.; et al. Epigenetic regulation of adaptive responses of forest tree species to the environment. Ecol. Evol. 2013, 3, 399–415. [Google Scholar] [CrossRef]
- Saze, H. Epigenetic memory transmission through mitosis and meiosis in plants. Semin. Cell Dev. Biol. 2008, 19, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Auler, P.A.; do Amaral, M.N.; Rodrigues, G.d.S.; Benitez, L.C.; da Maia, L.C.; Souza, G.M.; Braga, E.J.B. Molecular responses to recurrent drought in two contrasting rice genotypes. Planta 2017, 246, 899–914. [Google Scholar] [CrossRef] [PubMed]
- Yaish, M.W.; Al-Lawati, A.; Al-Harrasi, I.; Patankar, H.V. Genome-wide DNA Methylation analysis in response to salinity in the model plant caliph medic (Medicago truncatula). BMC Genom. 2018, 19, 78. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lang, C.; Wu, Y.; Meng, D.; Yang, T.; Li, D.; Jin, T.; Zhou, X. ROS1-mediated decrease in DNA methylation and increase in expression of defense genes and stress response genes in Arabidopsis thaliana due to abiotic stresses. BMC Plant Biol. 2022, 22, 104. [Google Scholar] [CrossRef]
- Zhang, J.; Colot, V.; Loudet, O.; Quadrana, L.; Tisné, S.; Bach, L.; Martin, A.; Jiménez-Gómez, J.M.; Gilbault, E.; Silveira, A.B.; et al. Mild drought in the vegetative stage induces phenotypic, gene expression, and DNA methylation plasticity in Arabidopsis but no transgenerational effects. J. Exp. Bot. 2020, 71, 3588–3602. [Google Scholar] [CrossRef]
- Lu, X.; Wang, X.; Chen, X.; Shu, N.; Wang, J.; Wang, D.; Wang, S.; Fan, W.; Guo, L.; Ye, W.; et al. Single-base resolution methylomes of upland cotton (Gossypium hirsutum L.) reveal epigenome modifications in response to drought stress. BMC Genom. 2017, 18, 207. [Google Scholar] [CrossRef]
- Li, R.; Hu, F.; Li, B.; Zhang, Y.; Chen, M.; Fan, T.; Wang, T. Whole genome bisulfite sequencing methylome analysis of mulberry (Morus alba) reveals epigenome modifications in response to drought stress. Sci. Rep. 2020, 10, 8013. [Google Scholar] [CrossRef]
- Sun, M.; Yang, Z.; Liu, L.; Duan, L. DNA Methylation in Plant Responses and Adaption to Abiotic Stresses. Int. J. Mol. Sci. 2022, 23, 6910. [Google Scholar] [CrossRef]
- Wang, W.; Qin, Q.; Sun, F.; Wang, Y.; Xu, D.; Li, Z.; Fu, B. Genome-Wide Differences in DNA Methylation Changes in Two Contrasting Rice Genotypes in Response to Drought Conditions. Front. Plant Sci. 2016, 7, 1675. [Google Scholar] [CrossRef]
- Tang, X.-M.; Tao, X.; Wang, Y.; Ma, D.-W.; Li, D.; Yang, H.; Ma, X.-R. Analysis of DNA methylation of perennial ryegrass under drought using the methylation-sensitive amplification polymorphism (MSAP) technique. Mol. Genet. Genom. 2014, 289, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Abid, G.; Mingeot, D.; Muhovski, Y.; Mergeai, G.; Aouida, M.; Abdelkarim, S.; Aroua, I.; El Ayed, M.; M’hamdi, M.; Sassi, K.; et al. Analysis of DNA methylation patterns associated with drought stress response in faba bean (Vicia faba L.) using methylation-sensitive amplification polymorphism (MSAP). Environ. Exp. Bot. 2017, 142, 34–44. [Google Scholar] [CrossRef]
- Neves, D.M.; Almeida, L.A.d.H.; Santana-Vieira, D.D.S.; Freschi, L.; Ferreira, C.F.; Soares Filho, W.d.S.; Costa, M.G.C.; Micheli, F.; Coelho Filho, M.A.; Gesteira, A.d.S. Recurrent water deficit causes epigenetic and hormonal changes in citrus plants. Sci. Rep. 2017, 7, 13684. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Huang, F.; Qin, Q.; Zhao, X.; Li, Z.; Fu, B. Comparative analysis of DNA methylation changes in two rice genotypes under salt stress and subsequent recovery. Biochem. Bioph. Res. Commun. 2015, 465, 790–796. [Google Scholar] [CrossRef]
- Wang, W.-S.; Pan, Y.-J.; Zhao, X.-Q.; Dwivedi, D.; Zhu, L.-H.; Ali, J.; Fu, B.-Y.; Li, Z.-K. Drought-induced site-specific DNA methylation and its association with drought tolerance in rice (Oryza sativa L.). J. Exp. Bot. 2011, 62, 1951–1960. [Google Scholar] [CrossRef]
- Gayacharan; Joel, A.J. Epigenetic responses to drought stress in rice (Oryza sativa L.). Physiol. Mol. Biol. Plants 2013, 19, 379–387. [Google Scholar] [CrossRef]
- Cao, Q.; Huang, L.; Li, J.; Qu, P.; Tao, P.; Crabbe, M.J.C.; Zhang, T.; Qiao, Q. Integrated transcriptome and methylome analyses reveal the molecular regulation of drought stress in wild strawberry (Fragaria nilgerrensis). BMC Plant Biol. 2022, 22, 613. [Google Scholar] [CrossRef]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; van Nocker, S.; Ma, F.; Guan, Q. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S.; et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012, 13, 30. [Google Scholar] [CrossRef]
- Niu, C.; Jiang, L.; Cao, F.; Liu, C.; Guo, J.; Zhang, Z.; Yue, Q.; Hou, N.; Liu, Z.; Li, X.; et al. Methylation of a MITE insertion in the MdRFNR1-1 promoter is positively associated with its allelic expression in apple in response to drought stress. Plant Cell 2022, 34, 3983–4006. [Google Scholar] [CrossRef]
- López, M.-E.; Roquis, D.; Becker, C.; Denoyes, B.; Bucher, E. DNA methylation dynamics during stress response in woodland strawberry (Fragaria vesca). Hortic. Res. 2022, 9, uhac174. [Google Scholar] [CrossRef] [PubMed]
- Virlouvet, L.; Avenson, T.J.; Du, Q.; Zhang, C.; Liu, N.; Fromm, M.; Avramova, Z.; Russo, S.E. Dehydration Stress Memory: Gene Networks Linked to Physiological Responses During Repeated Stresses of Zea mays. Front. Plant Sci. 2018, 9, 1058. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Vu, N.T.; Cheong, J.-J. Transcriptional Stress Memory and Transgenerational Inheritance of Drought Tolerance in Plants. Int. J. Mol. Sci. 2022, 23, 12918. [Google Scholar] [CrossRef]
- Szabados, L.; Savouré, A. Proline: A multifunctional amino acid. Trends Plant Sci. 2010, 15, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Hayat, S.; Hayat, Q.; Alyemeni, M.N.; Wani, A.S.; Pichtel, J.; Ahmad, A. Role of proline under changing environments. Plant Signal. Behav. 2012, 7, 1456–1466. [Google Scholar] [CrossRef]
- Parvaiz, A.; Satyawati, S. Salt stress and phyto-biochemical responses of plants—A review. Plant Soil Environ. 2008, 54, 89–99. [Google Scholar] [CrossRef]
- Li, P.; Yang, H.; Wang, L.; Liu, H.; Huo, H.; Zhang, C.; Liu, A.; Zhu, A.; Hu, J.; Lin, Y.; et al. Physiological and Transcriptome Analyses Reveal Short-Term Responses and Formation of Memory Under Drought Stress in Rice. Front. Genet. 2019, 10, 55. [Google Scholar] [CrossRef]
- Feng, X.J.; Li, J.R.; Qi, S.L.; Lin, Q.F.; Jin, J.B.; Hua, X.J. Light affects salt stress-induced transcriptional memory of P5CS1 in Arabidopsis. Proc. Natl. Acad. Sci. USA 2016, 113, E8335–E8343. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J. Salt and Drought Stress Signal Transduction in Plants. Annu. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef]
- Ullah, A.; Manghwar, H.; Shaban, M.; Khan, A.H.; Akbar, A.; Ali, U.; Ali, E.; Fahad, S. Phytohormones enhanced drought tolerance in plants: A coping strategy. Environ. Sci. Pollut. Res. 2018, 25, 33103–33118. [Google Scholar] [CrossRef]
- Guilfoyle, T.; Hagen, G.; Dong, T.; Park, Y.; Hwang, I. Abscisic acid: Biosynthesis, inactivation, homoeostasis and signalling. Essays Biochem. 2015, 58, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Fromm, M.; Avramova, Z. Multiple exposures to drought ‘train’ transcriptional responses in Arabidopsis. Nat. Commun. 2012, 3, 740. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Kim, D.H.; Hwang, I. ABA homeostasis and signaling involving multiple subcellular compartments and multiple receptors. Plant Cell Rep. 2013, 32, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Virlouvet, L.; Fromm, M. Physiological and transcriptional memory in guard cells during repetitive dehydration stress. New Phytol. 2014, 205, 596–607. [Google Scholar] [CrossRef]
- Li, Y.; Wang, M.; Guo, T.; Li, S.; Teng, K.; Dong, D.; Liu, Z.; Jia, C.; Chao, Y.; Han, L. Overexpression of abscisic acid-insensitive gene ABI4 from Medicago truncatula, which could interact with ABA2, improved plant cold tolerance mediated by ABA signaling. Front. Plant Sci. 2022, 13, 982715. [Google Scholar] [CrossRef]
- González-Guzmán, M.; Apostolova, N.; Bellés, J.M.; Barrero, J.M.; Piqueras, P.; Ponce, M.a.R.; Micol, J.L.; Serrano, R.n.; Rodríguez, P.L. The Short-Chain Alcohol Dehydrogenase ABA2 Catalyzes the Conversion of Xanthoxin to Abscisic Aldehyde. Plant Cell 2002, 14, 1833–1846. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zi, N.; Ren, W.; Guo, H.; Yuan, F.; Liu, Y.; Fry, E. DNA Methylation Participates in Drought Stress Memory and Response to Drought in Medicago ruthenica. Genes 2024, 15, 1286. https://doi.org/10.3390/genes15101286
Zi N, Ren W, Guo H, Yuan F, Liu Y, Fry E. DNA Methylation Participates in Drought Stress Memory and Response to Drought in Medicago ruthenica. Genes. 2024; 15(10):1286. https://doi.org/10.3390/genes15101286
Chicago/Turabian StyleZi, Na, Weibo Ren, Huiqin Guo, Feng Yuan, Yaling Liu, and Ellen Fry. 2024. "DNA Methylation Participates in Drought Stress Memory and Response to Drought in Medicago ruthenica" Genes 15, no. 10: 1286. https://doi.org/10.3390/genes15101286
APA StyleZi, N., Ren, W., Guo, H., Yuan, F., Liu, Y., & Fry, E. (2024). DNA Methylation Participates in Drought Stress Memory and Response to Drought in Medicago ruthenica. Genes, 15(10), 1286. https://doi.org/10.3390/genes15101286