
Clinical and Molecular Findings in Patients with Knobloch Syndrome 1: Case Series Report

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Cohort and Samples
2.2. Clinical Genetic Testing
2.2.1. DNA Purification
2.2.2. High-Throughput Sequencing
2.2.3. Sanger Sequencing
3. Results
3.1. Case 1. Family K
3.1.1. Medical History Data
3.1.2. Somatic and Neurologic Status
3.1.3. Ophthalmological Status
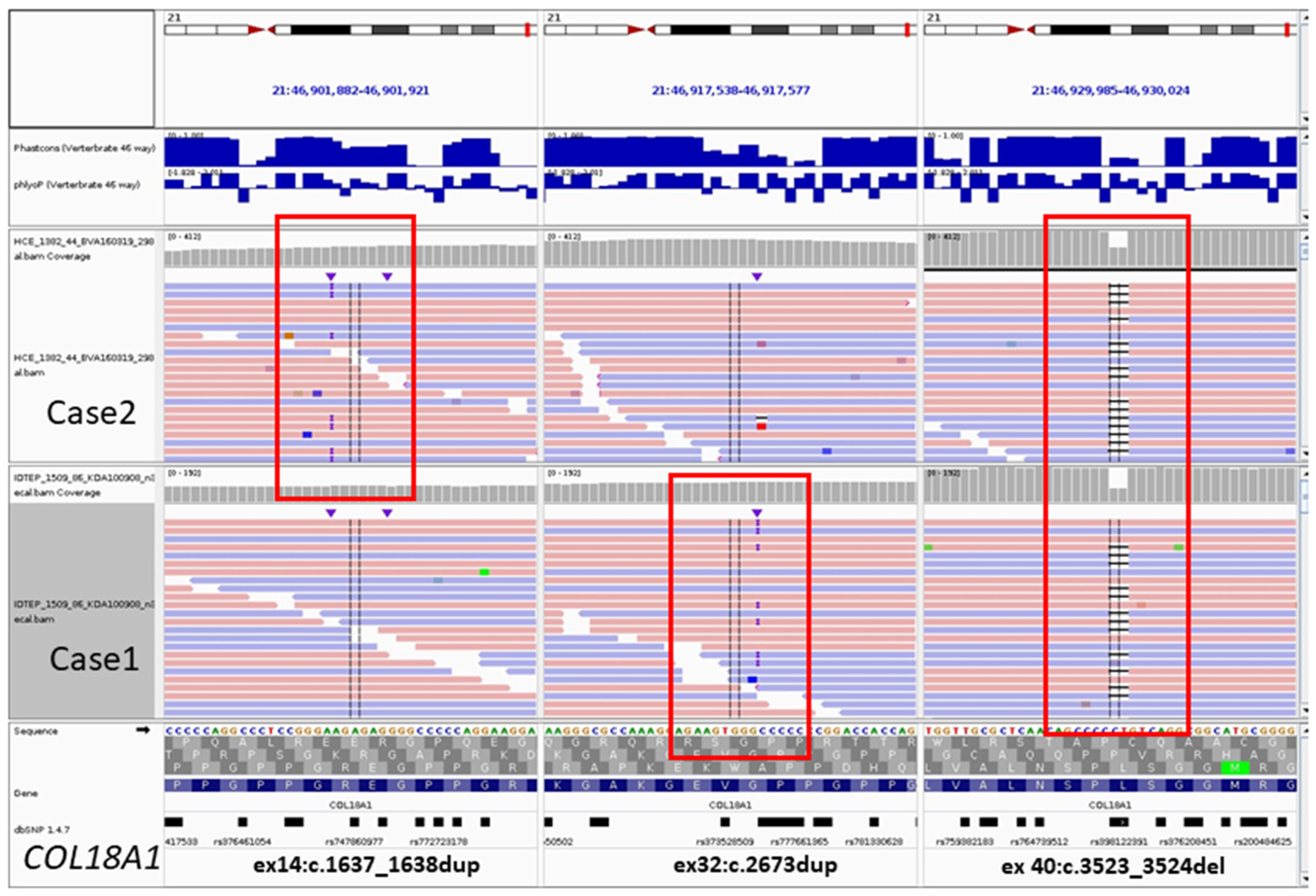
3.1.4. Genetic Testing
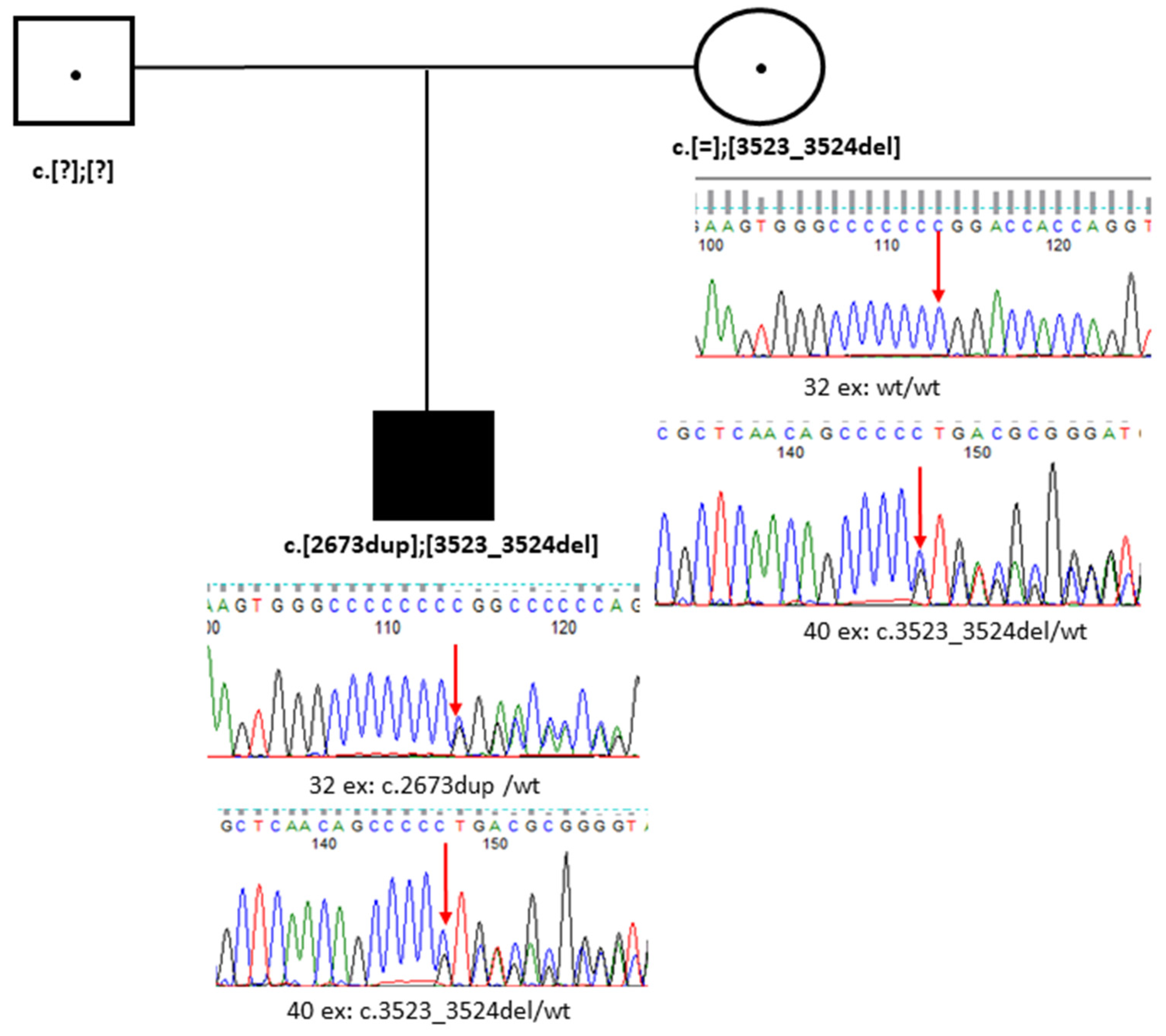
3.2. Case 2. Family B
3.2.1. Medical History Data
3.2.2. Ophthalmological Status
3.2.3. Genetic Testing
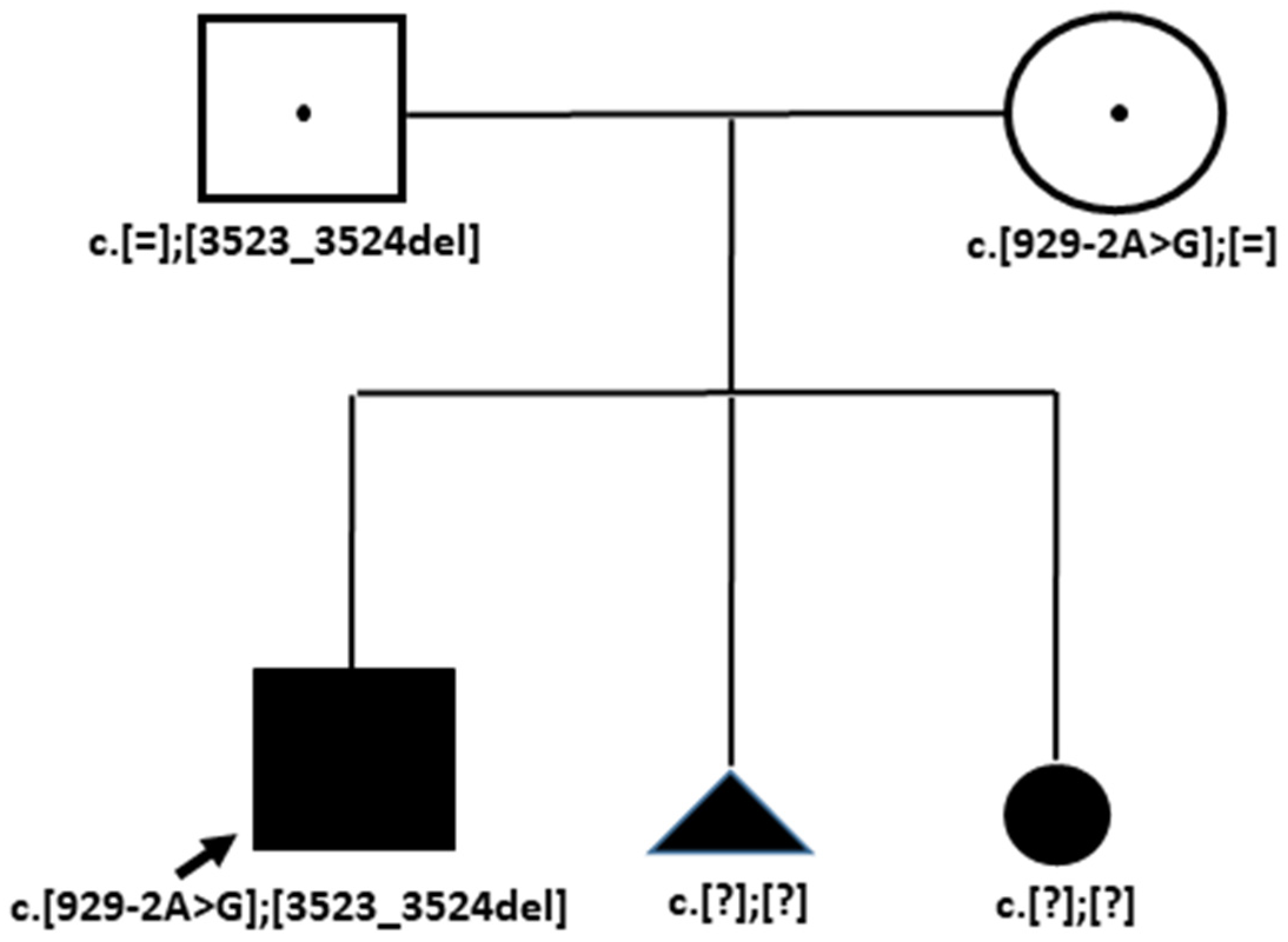
3.3. Case 3. Family OB
3.3.1. Medical History Data
3.3.2. Genetic Testing
4. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features/Patients | Patient 1 [24] | KS4.1 [12] | KS4.2 [12] | KS5 [12] | Case K (This Study) | Case B (This Study) | Case OB [10] |
|---|---|---|---|---|---|---|---|
| COL18A1 genotypes | c.2960_2969 dup | c.1238insA | c.1238insA | c.2105delC | c.2673dup | c.1637_1638 dup | c.929–2A>G |
| c.3523_3524 delCT | c.3523_3524 delCT | c.3523_3524 delCT | c.3523_3524 delCT | c.3523_3524 delCT | c.3523_3524 delCT | c.3523_3524 delCT | |
| SEX | F | F | M | M | M | F | M |
| AGE | 2 y.o. | 21 y.o. | 13 y.o. | 6 y.o. | 14 y.o. | 3 y.o. | 3 y.o. |
| Myopia magna | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Visual acuity (with optical correction) | 0.9 logmar and less than 1.3 logmar Decimal 0.125 и 0.005 | Blind (at age 5) | 20/200 best corrected (both eyes) | Blind (at age 1 year) | Low visual acuity | Low visual acuity | Low visual acuity |
| Nistagmus | Yes | n/m 1 | n/m | n/m | Yes | No | Yes |
| Eye fundus, retinal defect | Irregular macular region in the right fundus and atrophic lesion in the left fundus, thin retina with visible choroidal vessels | Retinal detachment, vitreoretinal degeneration | Retinal detachment, vitreoretinal degeneration | Retinal detachment | Tessellated fundus, thin retina with visible choroidal vessels, atrophic macula and optic nerve | Tessellated fundus, thin retina with visible choroidal vessels | Atrophic choroidea and optic nerve, tessellated fundus |
| Macula functionality | ERG showed the presence of scotopic and photopic responses, but severely reduced amplitudes and delayed latencies | n/m | n/m | n/m | Weakened reflex from the macula, retinal rod and cone dysfunction, trichromasia, and decreased subnormal ERG | Weakened reflex from the macula | n/m |
| Anterior segment | No | n/m | n/m | n/m | Atrophic iris, ectopia lentis, cataract | No | No |
| Occipital defect | Meningoencephalocele | Encephalocele | Encephalocele | Encephalocele | Cranial hernia | Dermoid cyst | Small posterior cranial fossa cyst |
| Additional connective tissue displasia features | n/m | No | No | No | Asthenic body constitution, funnel chest deformity, thin skin, joint hypermobility, blue sclera, midface hypoplasia | No | Asthenic body constitution, funnel chest deformity, thin skin, joint hypermobility, blue sclera, midface hypoplasia |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patil, S.J.; Pande, S.; Matalia, J.; Bhat, V.; Kekatpure, M.; Girisha, K.M. Knobloch syndrome in siblings with posterior fossa malformations along with cerebellar midline cleft abnormality caused by biallelic col18a1 mutation: Case-based review. J. Pediatr. Genet. 2023, 12, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Pufe, T.; Petersen, W.J.; Miosge, N.; Goldring, M.B.; Mentlein, R.; Varoga, D.J.; Tillmann, B.N. Endostatin/collagen xviii–an inhibitor of angiogenesis–is expressed in cartilage and fibrocartilage. Matrix Biol. 2004, 23, 267–276. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Zoeller, J.J.; Nystrom, A. Basement membrane proteoglycans: Modulators par excellence of cancer growth and angiogenesis. Mol. Cells 2009, 27, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Kliemann, S.E.; Waetge, R.T.; Suzuki, O.T.; Passos-Bueno, M.R.; Rosemberg, S. Evidence of neuronal migration disorders in knobloch syndrome: Clinical and molecular analysis of two novel families. Am. J. Med. Genet. A 2003, 119A, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Levinger, N.; Hendler, K.; Banin, E.; Hanany, M.; Kimchi, A.; Mechoulam, H.; Meiner, V.; Parag, Y.; Sharon, D.; Macarov, M.; et al. Variable phenotype of knobloch syndrome due to biallelic col18a1 mutations in children. Eur. J. Ophthalmol. 2021, 31, 3349–3354. [Google Scholar] [CrossRef] [PubMed]
- Hull, S.; Arno, G.; Ku, C.A.; Ge, Z.; Waseem, N.; Chandra, A.; Webster, A.R.; Robson, A.G.; Michaelides, M.; Weleber, R.G.; et al. Molecular and clinical findings in patients with knobloch syndrome. JAMA Ophthalmol. 2016, 134, 753–762. [Google Scholar] [CrossRef]
- Richards, A.J.; Meredith, S.; Poulson, A.; Bearcroft, P.; Crossland, G.; Baguley, D.M.; Scott, J.D.; Snead, M.P. A novel mutation of col2a1 resulting in dominantly inherited rhegmatogenous retinal detachment. Invest. Ophthalmol. Vis. Sci. 2005, 46, 663–668. [Google Scholar] [CrossRef]
- Rudenskaya, G.E.; Marakhonov, A.V.; Shchagina, O.A.; Lozier, E.R.; Dadali, E.L.; Akimova, I.A.; Petrova, N.V.; Konovalov, F.A. Ataxia with oculomotor apraxia type 4 with pnkp common “portuguese” and novel mutations in two belarusian families. J. Pediatr. Genet. 2019, 8, 58–62. [Google Scholar] [CrossRef]
- Kohler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The human phenotype ontology in 2021. Nucleic. Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Khlebnikova, O.V.; Dadali, E.L.; Bessonova, L.A.; Konovalov, F.A. Clinical-genetic features of the knobloch syndrome in the russian family. Russ. Pediatr. Ophthalmol. (In Russian). 2018, 13, 109–112. [Google Scholar] [CrossRef]
- Suzuki, O.; Kague, E.; Bagatini, K.; Tu, H.; Heljasvaara, R.; Carvalhaes, L.; Gava, E.; de Oliveira, G.; Godoi, P.; Oliva, G.; et al. Novel pathogenic mutations and skin biopsy analysis in knobloch syndrome. Mol. Vis. 2009, 15, 801–809. [Google Scholar] [PubMed]
- Suzuki, O.T.; Sertie, A.L.; Der Kaloustian, V.M.; Kok, F.; Carpenter, M.; Murray, J.; Czeizel, A.E.; Kliemann, S.E.; Rosemberg, S.; Monteiro, M.; et al. Molecular analysis of collagen xviii reveals novel mutations, presence of a third isoform, and possible genetic heterogeneity in knobloch syndrome. Am. J. Hum. Genet. 2002, 71, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Coban-Akdemir, Z.; White, J.J.; Song, X.; Jhangiani, S.N.; Fatih, J.M.; Gambin, T.; Bayram, Y.; Chinn, I.K.; Karaca, E.; Punetha, J.; et al. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-function alleles. Am. J. Hum. Genet. 2018, 103, 171–187. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting splicing from primary sequence with deep learning. Cell 2019, 176, 535–548.e524. [Google Scholar] [CrossRef]
- Passos-Bueno, M.R.; Suzuki, O.T.; Armelin-Correa, L.M.; Sertie, A.L.; Errera, F.I.; Bagatini, K.; Kok, F.; Leite, K.R. Mutations in collagen 18a1 and their relevance to the human phenotype. An. Acad. Bras. Cienc. 2006, 78, 123–131. [Google Scholar] [CrossRef]
- Fukai, N.; Eklund, L.; Marneros, A.G.; Oh, S.P.; Keene, D.R.; Tamarkin, L.; Niemela, M.; Ilves, M.; Li, E.; Pihlajaniemi, T.; et al. Lack of collagen xviii/endostatin results in eye abnormalities. EMBO J. 2002, 21, 1535–1544. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The human gene mutation database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef]
- Jiang, Z.; Sun, W.; Zhang, Q.; Wang, P. Generation of an induced pluripotent stem cell line (ZSZOCi001-A) from a patient with Knob-loch syndrome caused by biallelic mutations in the gene COL18A1. Stem. Cell Res. 2023, 70, 103131. [Google Scholar] [CrossRef]
- Ogreden, T.A.; Erdogan, G. Two patients with knobloch syndrome due to mutation in col8a1 gene: Case report and review of the literature. BMC Ophthalmol. 2024, 24, 149. [Google Scholar] [CrossRef] [PubMed]
- Caglayan, A.O.; Baranoski, J.F.; Aktar, F.; Han, W.; Tuysuz, B.; Guzel, A.; Guclu, B.; Kaymakcalan, H.; Aktekin, B.; Akgumus, G.T.; et al. Brain malformations associated with knobloch syndrome–review of literature, expanding clinical spectrum, and identification of novel mutations. Pediatr. Neurol. 2014, 51, 806–813.e808. [Google Scholar] [CrossRef] [PubMed]
- Keren, B.; Suzuki, O.T.; Gerard-Blanluet, M.; Bremond-Gignac, D.; Elmaleh, M.; Titomanlio, L.; Delezoide, A.L.; Passos-Bueno, M.R.; Verloes, A. Cns malformations in knobloch syndrome with splice mutation in col18a1 gene. Am. J. Med. Genet. A 2007, 143A, 1514–1518. [Google Scholar] [CrossRef]
- Mayer, A.K.; Balousha, G.; Sharkia, R.; Mahajnah, M.; Ayesh, S.; Schulze, M.; Buchert, R.; Zobor, D.; Azem, A.; Schols, L.; et al. Unraveling the genetic cause of hereditary ophthalmic disorders in arab societies from israel and the palestinian authority. Eur. J. Hum. Genet. 2020, 28, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Balikova, I.; Sanak, N.S.; Fanny, D.; Smits, G.; Soblet, J.; de Baere, E.; Cordonnier, M. Three cases of molecularly confirmed knobloch syndrome. Ophthalmic Genet. 2020, 41, 83–87. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Scopes, G.; Lee, P.; Houlden, H. Homozygosity mapping through whole genome analysis identifies a col18a1 mutation in an indian family presenting with an autosomal recessive neurological disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2009, 150B, 993–997. [Google Scholar] [CrossRef]
- Zhang, L.S.; Li, H.B.; Zeng, J.; Yang, Y.; Ding, C. Knobloch syndrome caused by homozygous frameshift mutation of the col18a1 gene in a chinese pedigree. Int. J. Ophthalmol. 2018, 11, 918–922. [Google Scholar]
- Zech, T.J.; Wolf, A.; Hector, M.; Bischoff-Kont, I.; Krishnathas, G.M.; Kuntschar, S.; Schmid, T.; Bracher, F.; Langmann, T.; Furst, R. 2-desaza-annomontine (c81) impedes angiogenesis through reduced vegfr2 expression derived from inhibition of cdc2-like kinases. Angiogenesis 2024, 27, 245–272. [Google Scholar] [CrossRef]
- Xu, M.; Shen, Y.M.; Han, X.Y.; Liu, C.; Jiang, Q.; Cao, X.; Yan, B. “One stone and two birds” strategy to treat neovascular age-related macular degeneration by a novel retinoid drug, eye-101. Exp. Eye Res. 2023, 227, 109385. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, L.; Dong, C.; Yang, X.; Wang, J.; Zhang, X.; Yuan, Y.; Dai, J.; Huang, J.; Yuan, F. Microrna-376b-3p suppresses choroidal neovascularization by regulating glutaminolysis in endothelial cells. Investig. Ophthalmol. Vis. Sci. 2023, 64, 22. [Google Scholar] [CrossRef]
- Gatseva, A.; Sin, Y.Y.; Brezzo, G.; Van Agtmael, T. Basement membrane collagens and disease mechanisms. Essays Biochem. 2019, 63, 297–312. [Google Scholar]
- Egea, G.; Jimenez-Altayo, F.; Campuzano, V. Reactive oxygen species and oxidative stress in the pathogenesis and progression of genetic diseases of the connective tissue. Antioxidants 2020, 9, 1013. [Google Scholar] [CrossRef] [PubMed]
- Heljasvaara, R.; Aikio, M.; Ruotsalainen, H.; Pihlajaniemi, T. Collagen xviii in tissue homeostasis and dysregulation - lessons learned from model organisms and human patients. Matrix Biol. 2017, 57-58, 55–75. [Google Scholar] [CrossRef] [PubMed]
- Mettu, P.S.; Allingham, M.J.; Cousins, S.W. Incomplete response to anti-vegf therapy in neovascular amd: Exploring disease mechanisms and therapeutic opportunities. Prog. Retin. Eye Res. 2021, 82, 100906. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.; Lee, D.; Negishi, K.; Tsubota, K.; Kurihara, T. Establishment of an in vitro choroid complex system for vascular response screening. Sci. Rep. 2024, 14, 16129. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Shao, Z.; Cakir, B.; Kotoda, Y.; Fu, Z.; Smith, L.E.H. An ex vivo choroid sprouting assay of ocular microvascular angiogenesis. J. Vis. Exp. 2020, 162. [Google Scholar]
- Fomo, K.N.; Perumal, N.; Manicam, C.; Pfeiffer, N.; Grus, F.H. Neuroretinal cell culture model as a tool for the development of new therapeutic approaches for oxidative stress-induced ocular diseases, with a focus on glaucoma. Cells 2024, 13, 775. [Google Scholar] [CrossRef]


| Family with a Patient | Case 1/Family K | Case 2/Family B | Case 3/Family OB |
|---|---|---|---|
| Sex | M | F | M |
| Age | 14 y.o. | 3 y.o. | 3 y.o. |
| Retinal detechment | No | No | No |
| Myopia (diopter—D) | >10–13 D, compound myopic astigmatism | >6 D, compound myopic astigmatism | 18 D, compound myopic astigmatism |
| Strabismus | Nonconcomitant strabismus | No | n/m |
| Lens subluxation | No | No | n/m |
| Cataract | Yes | No | No |
| Vitreoretinal destruction | Yes, central eyeball fibers | n/m 1 | n/m |
| Visual acuty | 0.05 | 0.1 | 0.09 |
| Choroid atrophy | Yes | Yes | Yes |
| Optic nerve atrophy | Optic nerve partial atrophy | Pale-pink with clear boundaries | Decolorized optic disk |
| Macula reflex abnormality | Weak reflex | Weak/intermittent reflex | n/m |
| Nistagmus | Yes | No | Yes |
| Fundus pigment dissociation | Yes | Yes | Yes |
| Occipital skull defect | Osseous tissue defect 3.3 mm, meningocele 1.6 × 0.95 mm | Dermoid cyst, occipital hemangioma 0.5 cm | Small posterior cranial fossa cyst |
| Neurological phenotype | No | No | No |
| Psychomotor development | No | No | No |
| Asthenic constitution | Yes | n/m | Yes |
| Pectus excavatum (funnel chest) | Yes | Yes | Yes |
| Thin skin | n/m | n/m | Yes |
| Joint hypermobility | Yes | n/m | Yes |
| Blue eye sclera | n/m | n/m | Yes |
| Face deformity | Elongated face, low forehead, downward palpebral slant, massive chin, large nasal bridge | n/m | Hypoplastic midface |
| Genotype NM_001379500.1(COL18A1) | c.[2673dup]; [3523_3524del] | c.[3523_3524del]; [1637_1638dup] | c.[3523_3524del]; [929-2A>G] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasilyeva, T.; Kadyshev, V.; Khalanskaya, O.; Kuznetsova, S.; Ionova, S.; Marakhonov, A.; Zinchenko, R. Clinical and Molecular Findings in Patients with Knobloch Syndrome 1: Case Series Report. Genes 2024, 15, 1295. https://doi.org/10.3390/genes15101295
Vasilyeva T, Kadyshev V, Khalanskaya O, Kuznetsova S, Ionova S, Marakhonov A, Zinchenko R. Clinical and Molecular Findings in Patients with Knobloch Syndrome 1: Case Series Report. Genes. 2024; 15(10):1295. https://doi.org/10.3390/genes15101295
Chicago/Turabian StyleVasilyeva, Tatyana, Vitaly Kadyshev, Olga Khalanskaya, Svetlana Kuznetsova, Sofya Ionova, Andrey Marakhonov, and Rena Zinchenko. 2024. "Clinical and Molecular Findings in Patients with Knobloch Syndrome 1: Case Series Report" Genes 15, no. 10: 1295. https://doi.org/10.3390/genes15101295