
Genetic Insights into the Giant Keyhole Limpet (Megathura crenulata), an Eastern Pacific Coastal Endemic: Complete Mitogenome, Phylogenetics, Phylogeography, and Historical Demography
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling for Mitogenome Analysis
2.2. Bioinformatic Analysis
2.3. Primer Design to Circularize the Mitogenome
2.4. Mitogenome Annotation
2.5. Phylogenetic Analyses of Marine Gastropods
2.6. Sampling for Mitochondrial Genetic Diversity and Structure Analysis
2.7. Diversity and Genetic Structure Analysis
2.8. Historic Demographic Analyses
3. Results
3.1. Mitogenome Content and Organization
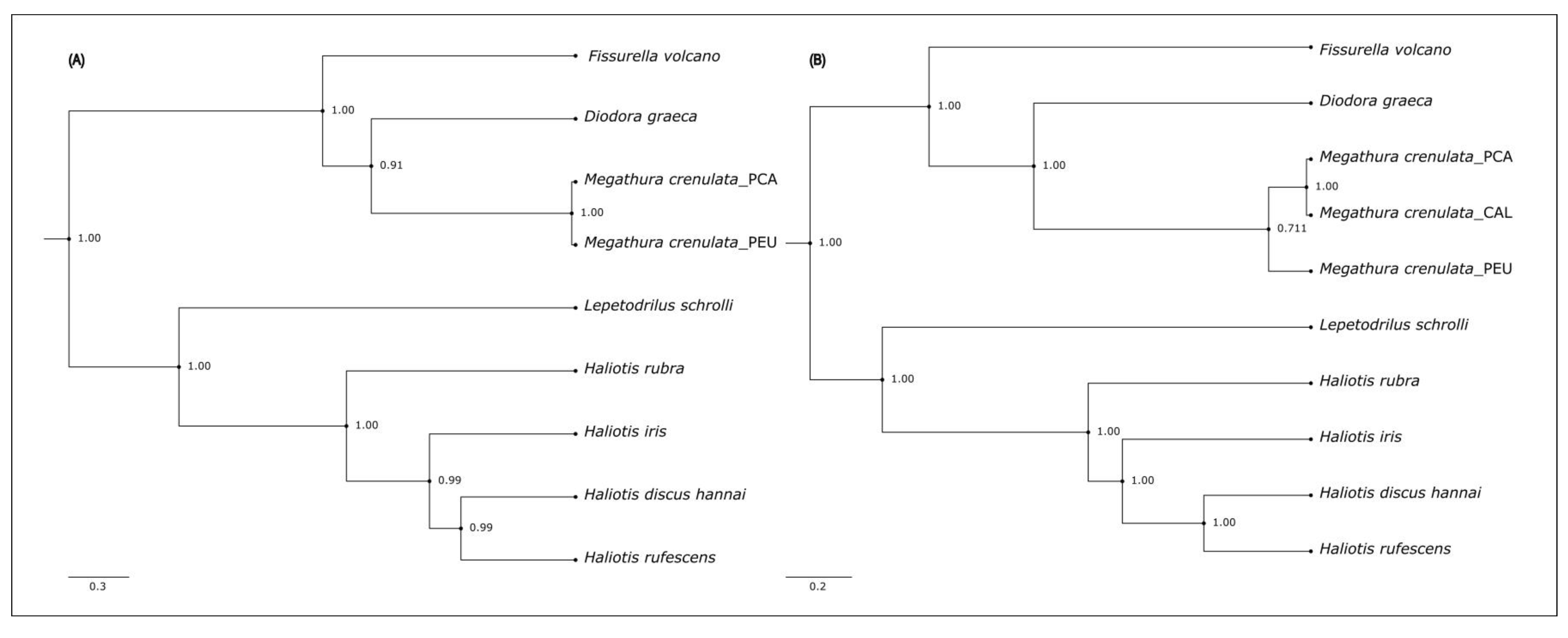
3.2. Phylogenetic Analysis
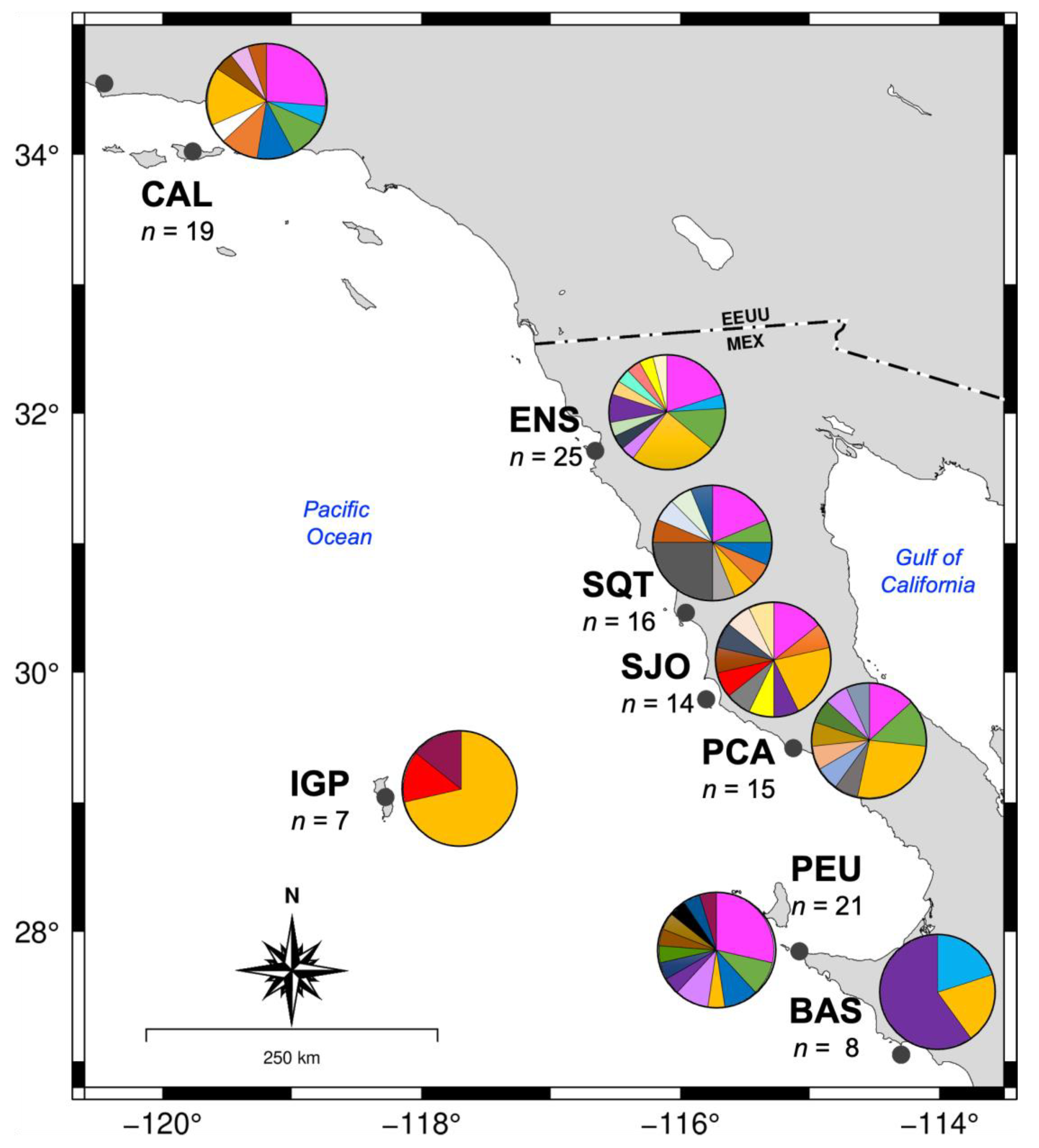
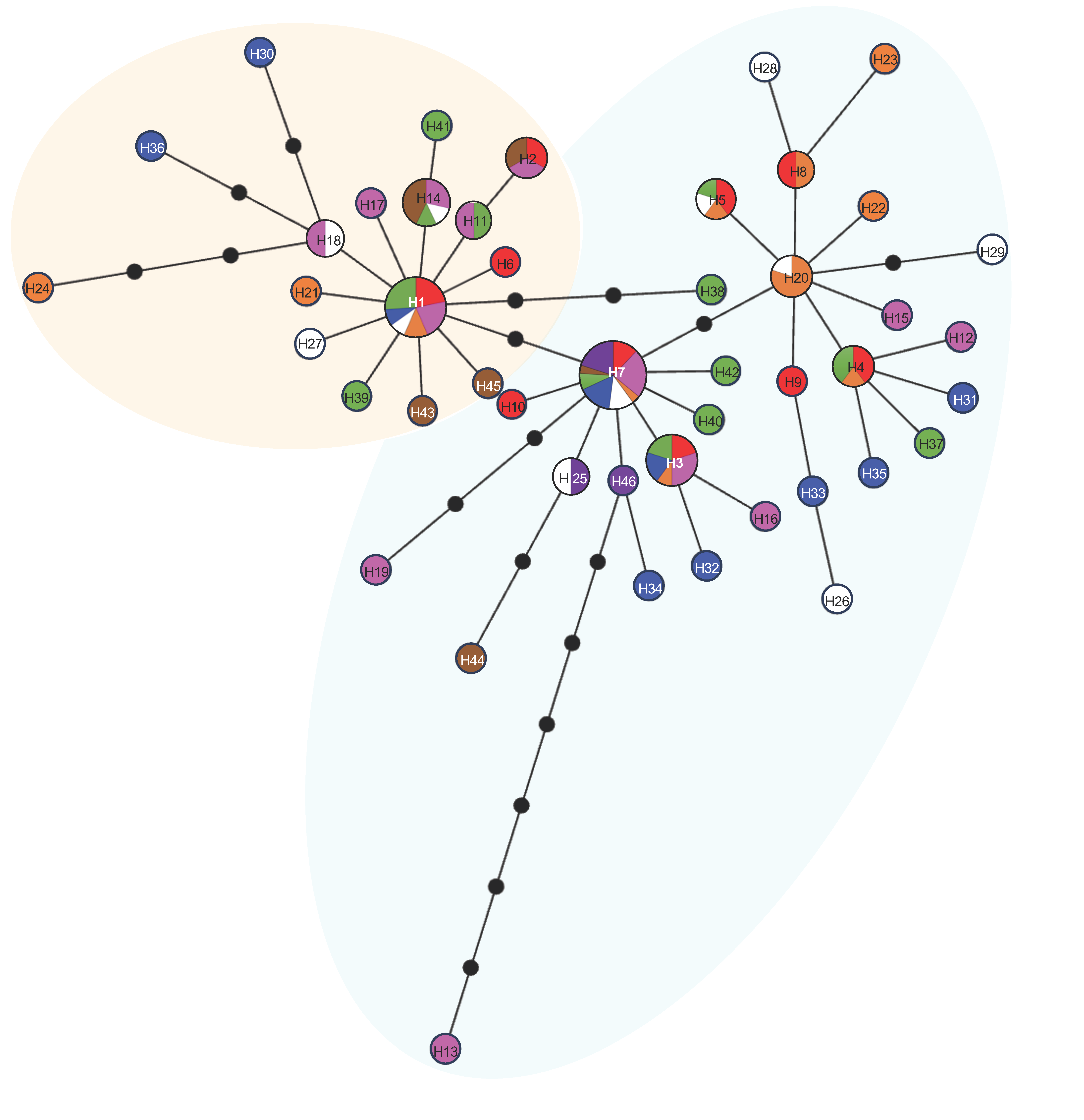
3.3. Genetic Diversity and Population Structure
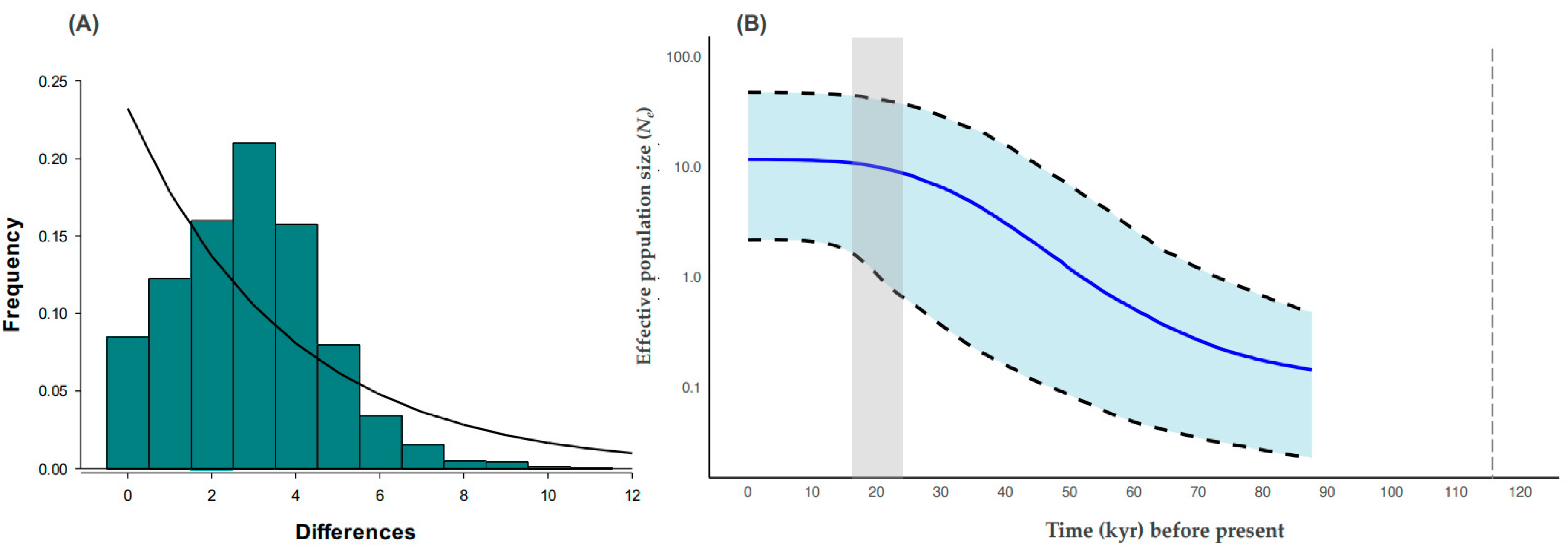
3.4. Historic Demography
4. Discussion
4.1. Mitogenome Content and Organization
4.2. Phylogenetic Analysis
4.3. Genetic Diversity and Population Structure
4.4. Historic Demography
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sowerby, G.B. A Catalogue of the Shells Contained in the Collection of the Late Earl of Tankerville: Arranged According to the Lamarckian Conchological System; Together with an Appendix Containing Descriptions of Many New Species; EJ Stirling: London, UK, 1825. [Google Scholar]
- Torres-Moye, G. Mainland and Island Kelp Forests in Northern Baja California, México: Subtidal Community Structure, Dynamics, and Connectivity for the Design of Marine Protected Areas. Ph.D. Thesis, University of California, Davis, CA, USA, 2012. [Google Scholar]
- Mau, A.; Jha, R. Aquaculture of two commercially important mollusks (abalone and limpet): Existing knowledge and futures prospects. Rev. Aquac. 2018, 10, 611–625. [Google Scholar] [CrossRef]
- Miller, C.; Robbins, R. Chemical analysis and vitamin assays of opihi, the Hawaiian limpet. Philipp. J. Sci. 1940, 71, 141–163. Available online: https://www.cabidigitallibrary.org/doi/full/10.5555/19401401307 (accessed on 1 January 2020).
- Oakes, F.R.; Mctee, S.; Culver, J.; Morse, D. El efecto del cautiverio y la dieta sobre las proporciones de isoformas de KLH en Megathura crenulata. Bio. Comp. Y Fisiol. Parte A Fisiol. Mol. E Integ. 2004, 138, 169–173. [Google Scholar] [CrossRef]
- Swerdlow, R.D.; Ebert, R.F.; Lee, P.; Bonaventura, C.; Miller, K.I. Keyhole limpet hemocyanin: Structural and functional characterization of two different subunits and multimers. CBPB 1996, 113, 537–548. [Google Scholar] [CrossRef]
- Del Campo, M.; Arancibia, S.; Nova, E.; Salazar, F.; González, A.; Moltedo, B.; De-Loannes, P.; Ferreira, J.; Manubens, A.; Becker, M.I. Hemocianinas, una herramiento inmunológica de la biomedicina actual. Rev. Méd. Chile 2011, 139, 236–246. [Google Scholar] [PubMed]
- Becker, M.I.; Arancibia, S.; Salazar, F.; Del Campo, M.; De-Loannes, A. Mollusk Hemocyanin’s as natural immunostimulants in biomedical applications. In Immune Response Activation; Duc, G.H.T., Ed.; IntechOpen: Croatia, Rijeka, 2014. [Google Scholar]
- Markl, J.; Savel-Niemann, A.; Wegener-Strake, A.; Soling, M.; Schneider, A.; Gebauer, W.; Harris, J.R. The Role of two distinct subunit types in the architecture of Keyhole Limpet Hemocyanin (KLH). Naturwissenschaften 1991, 78, 512–514. [Google Scholar] [CrossRef]
- SEPESCA. Carta Estatal Pesquera De Baja California. 2015. Available online: https://www.bajacalifornia.gob.mx/Documentos/Sepesca/doctos/CARTA_ESTATAL_PESQUERA_2000-2018.pdf (accessed on 5 January 2024).
- Micheli, F.; Saenz-Arroyo, A.; Aalto, E.; Beas-Luna, R.; Boch, C.A.; Cárdenas, J.C.; Woodson, C.B. Social-ecological vulnerability to environmental extremes and adaptation pathways in small-scale fisheries of the southern California Current. Front. Mar. Sci. 2024, 11, 1322108. [Google Scholar] [CrossRef]
- Gómez-Valdes, M.M. Caracterización del Nivel Trófico de Megathura crenulata (Sowerby, 1825) Mediante el Uso de la Señal isotópica del δ13C y δ15N. Master’s Thesis, Centro de Investigaciones Biológicas del Noroeste, La Paz, Baja California, México, November 2012. [Google Scholar]
- Munasinghe, M.; Ågren, J.A. When and why are mitochondria paternally inherited? Curr. Opin. Genet. Dev. 2023, 80, 102053. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 767–780. [Google Scholar] [CrossRef]
- Qi, L.; Xu, B.; Kong, L.; Li, Q. Improved phylogenetic resolution within Neritidae (Gastropoda, Nertimorpha) with implications for the evolution of shell traits and habitat. Zool. Scr. 2023, 52, 46–57. [Google Scholar] [CrossRef]
- An, H.S.; Jee, Y.J.; Min, K.S.; Kim, B.L.; Han, S.J. Phylogenetic analysis of six species of pacific abalone (Haliotidae) based on DNA sequences of 16s rRNA and Cytochrome C Oxidase Subunit I mitochondrial genes. Mar. Biotech. 2005, 7, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Ghiselli, F.; Gomes-Dos-Santos, A.; Adema, C.M.; Lopes-Lima, M.; Sharbrough, J.; Boore, J.L. Molluscan mitochondrial genomes break the rules. Philos. Trans. R Soc. Lond. B Biol. Sci. 2021, 376, 20200159. [Google Scholar] [CrossRef] [PubMed]
- Uribe, J.E.; Sei, M.; Harasewych, M.G. The mitogenome of the sunken wood limpet Notocrater youngi: Insights into mitogenome evolution in Lepetellida (Gastropoda: Vetigastropoda). J. Molluscan Stud. 2022, 88, eyac009. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritish, E.F.; Maniatals, T. Molecular Cloning: A Laboratory Manual; CSHL Press: Cold Spring Harbor, NY, USA, 1989; p. 1626. [Google Scholar]
- Glenn, T.C.; Nilsen, R.A.; Kieran, T.J.; Sanders, J.G.; Bayona-Vásquez, N.J.; Finger, J.W.; Pierson, T.W.; Bentley, K.E.; Hoffberg, S.L.; Louha, S.; et al. Adapterama I: Universal stubs and primers for 384 unique dual-indexed or 147,456 combinatorially-indexed Illumina libraries (iTru & iNext). PeerJ 2019, 7, e7755. [Google Scholar] [CrossRef]
- Rohland, N.; Reich, D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 2012, 22, 939–946. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, L.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-New capabilities and interfaces. Nucleic Acids Res. 2012, 40, 115. [Google Scholar] [CrossRef]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Simison, W.B. Center for Comparative Genomics, California. San Francisco California, USA. mitochondrion Fissurella volcano (volcano limpet). 2012; Unpublished work. [Google Scholar]
- Fang, X.; Guo, Z.; Zhang, W.; Li, L.; Zhang, J.; Zhang, H.; Hou, X. National Center for Biotechnology. Bethesda, Maryland, USA. Mitochondrial Haliotis iris. 2023; Unpublished work. [Google Scholar]
- Maynard, B.T.; Kerr, L.J.; Mckiernan, J.M.; Jansen, E.S.; Hanna, P.J. Mitochondrial DNA sequence and gene organization in the Australian blacklip abalone Haliotis rubra (Leach). Mar. Biotech. 2005, 7, 645–658. [Google Scholar] [CrossRef]
- del Rio-Portilla, M.A.; Lafarga De La Cruz, F.; Vargas-Peralta, C.E.; Farfan, C. Centro de Investigación Científica y de Educación Siperior de Ensenada, Baja California, México. Complete mitochondrial genome of the red abalone, Haliotis rufescens Swainson, 1822: A mitogenome realignment of Haliotidae. 2018; Unpublished work. [Google Scholar]
- Ren, J.; Shen, X.; Liu, X. Institute of Oceanology, Qingdao, Shandong, China. Sequence analysis in the mitochondrial genome of the Pacific abalone Haliotis discus hannai (Gastropoda, Haliotidae) and exploitation of its application. 2008; Unpublished work. [Google Scholar]
- Santorum, J.M.; Darriba, D.; Taboada, G.L.; Posada, D. Jmodeltest. org: Selection of nucleotide substitution models on the cloud. Bioinformatics 2014, 30, 1310–1311. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Palumbi, S.; Martin, A.; Romano, S.; Mcmillan, W.O.; Stice, L.; Grabowski, G. The Simple Fools Guide to PCR; University of Hawaii: Honolulu, HI, USA, 2022. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-Del Barrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-García, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Rice, W.R. Analyzing tables of statistical tests. Evolution 1989, 43, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Sokal, R.; Rohlf, J.F. Biometry: The Principles and Practices of Statistics in Biological Research; W.H Freeman and Company: New York, NY, USA, 1969. [Google Scholar]
- Teacher, A.G.F.; Griffiths, D.J. HapStar: Automated haplotype network layout and visualization. Mol. Ecol. Res. 2011, 11, 151–153. [Google Scholar] [CrossRef]
- Ho, S.Y.W.; Shapiro, B. Skyline-plot methods for estimating demographic history from nucleotide sequences. Mol. Ecol. Resour. 2011, 11, 423–434. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. MBE 2005, 22, 1185–1192. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Drummond, A.J. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed]
- Wilke, T.; Schultheiß, R.; Albrecht, C. As time goes by a simple fool’s guide to molecular clock approaches in invertebrates. Am. Malacol. Bull. 2009, 27, 25–45. [Google Scholar] [CrossRef]
- Uribe-Arboleda, J.E. Mitogenómica y Filogenía de Linajes de Gasterópodos Altamente Diversificados (Vetigastropoda, Neritimorpha y Conoidea). Ph.D. Thesis, Universidad Autónoma de Madrid, Madrid, Spain, 2016. [Google Scholar]
- Robinson, N.A.; Hall, N.E.; Ross, E.M.; Cooke, I.R.; Shiel, B.P.; Robinson, A.J.; Strugnell, J.M. The complete mitochondrial genome of Haliotis laevigata (Gastropoda: Haliotidae) using MiSeq and HiSeq sequencing. Mitochondrial DNA Part A 2016, 27, 437–438. [Google Scholar] [CrossRef]
- Rui, G.; Shanpi, Z.; Leiming, C.; Xiaoqiang, Y.; Wei, W.; Xiaobin, J.; Zhelong, L. Sequencing and analysis of the complete mitochondrial genome of green abalone (Haliotis fulgens). Acta Oceanogr. 2023, 45, 64–78. [Google Scholar] [CrossRef]
- Grande, C.; Templado, J.; Zardoya, R. Evolution of gastropod mitochondrial genome arrangements. BMC Evol. Biol. 2008, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Eldem, V.; Balci, M.A. Mining NCBI Sequence Read Archive Database: An Untapped Source of Organelle Genomes for Taxonomic and Comparative Genomics Research. Diversity 2024, 16, 104. [Google Scholar] [CrossRef]
- Aguilera-Muñoz, F.; Lafarga-Cruz, F.; Gallardo-Escaráte, C. Molecular Analysis in Chilean commercial gastropods based on 16s rRNA, COX1 and ITS 1-5.8S RDNA-ITS2 sequences. Gayana 2009, 73, 17–27. [Google Scholar] [CrossRef]
- Grande, C.; Zardoya, R. Moluscos. Digital Csic. 2014. Available online: http://hdl.handle.net/10261/100133 (accessed on 1 June 2023).
- Breton, S.; Beaupre, H.D.; Stewart, D.T.; Hoeh, W.R.; Blier, P.U. The unusual system of doubly uniparental inheritance of mtDNA: Isn’t one enough? Trends Genet. 2007, 23, 465–474. [Google Scholar] [CrossRef]
- Skibinski, D.O.; Gallagher, C.; Beynon, C.M. Mitochondrial DNA inheritance. Nature 1994, 368, 817–818. [Google Scholar] [CrossRef]
- Zouros, E.; Ball, A.O.; Saavedra, C.; Freeman, K.R. Mitochondrial DNA inheritance. Nature 1994, 368, 818. [Google Scholar] [CrossRef]
- Cao, L.; Kenchington, E.; Zouros, E.; Rodakis, G.C. Evidence that the large noncoding sequence is the main control region of maternally and paternally transmitted mitochondrial genomes of the marine mussel (Mytilus spp.). Genetics 2004, 167, 835–850. [Google Scholar] [CrossRef]
- Giantsis, I.A.; Abatzopoulos, T.J.; Angelidis, P.; Apostolidis, A.P. Mitochondrial control region variability in Mytilus galloprovincialis populations from the Central-Eastern Mediterranean Sea. Int. J. Mol. Sci. 2014, 15, 11614–11625. [Google Scholar] [CrossRef]
- Van Wormhoudt, A.; Roussel, V.; Courtois, G.; Huchette, S. Mitochondrial DNA introgression in the European abalone Haliotis tuberculata tuberculata: Evidence for experimental mtDNA paternal inheritance and a natural hybrid sequence. Mar. Biotech. 2011, 13, 563–574. [Google Scholar] [CrossRef]
- Gusman, A.; Azuelos, C.; Breton, S. No evidence of sex-linked heteroplasmy or doubly-uniparental inheritance of mtDNA in five gastropod species. J. Molluscan Stud. 2017, 83, 119–122. [Google Scholar] [CrossRef]
- Cárdenas, L.; Castilla, J.C.; Viard, F. A phylogeographical analysis across three biogeographical provinces of the south-eastern Pacific: The case of the marine gastropod Concholepas Concholepas. J. Biogeogr. 2009, 36, 969–981. [Google Scholar] [CrossRef]
- Nuñez, J.D.; Fernández-Iriarte, P.J.; Ocampo, E.H.; Iudica, C.; Cledón, M. Deep phylogeographic divergence among populations of limpet Siphonaria lessoni on the east and west coasts of South America. Mar. Biol. 2015, 162, 595–605. [Google Scholar] [CrossRef]
- Klinbunga, S.; Pripue, P.; Khamnamtong, N.; Puanglarp, N.; Tassanakajon, A.; Jarayabhand, P.; Hirono, I.; Aoki, T.; Menasveta, P. Genetic diversity and molecular markers of the tropical abalone (Haliotis asinina) in Thailand. Mar. Biotech. 2003, 5, 505–517. [Google Scholar] [CrossRef]
- Grant, W.S.; Bowen, W. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. J. Hered. 1998, 89, 415–426. [Google Scholar] [CrossRef]
- Río-Portilla, M.A.; González-Avilés, J.G. Population genetics of the yellow abalone, Haliotis corrugata, in Cedros and San Benito Islands: A preliminary survey. J. Shellfish Res. 2001, 20, 765–770. [Google Scholar]
- Buenrostro, A.A.; Rodríguez, M.F.; Peraza, M.M.; Espinoza, M.L.A. Variaciones de período corto de la circulación anticiclónica en Bahía Sebastián Vizcaíno, BC. Cienc. Mar. 1995, 21, 201–223. [Google Scholar] [CrossRef]
- Armijo-De Vega, C. Crecimiento y Reproducción de la Lapa Megathura crenulata en la Bahía de Todos Santos Baja California, Mexico. Master’s Thesis, Universidad Autónoma de Baja California, Ensenada, BS, Mexico, February 1993. [Google Scholar]
- Mejía-Ruíz, P.; Perez-Enriquez, R.; Mares-Mayagoitia, J.A.; Valenzuela-Quiñonez, F. Population genomics reveals a mismatch between management and biological units in green abalone (Haliotis fulgens). PeerJ 2020, 8, e9722. [Google Scholar] [CrossRef]
- Mares-Mayagoitia, J.; De-Anda-Montañez, J.A.; Perez-Enriquez, R.; Mejía-Ruíz, P.; Gutiérrez-González, J.L.; Valenzuela-Quiñonez, F. Neutral and adaptive population structure of pink abalone (Haliotis corrugata): Fishery management implications. J. Mar. Sci. 2021, 78, 1909–1919. [Google Scholar] [CrossRef]
- Grant, W.S. Problems and cautions with sequence mismatch analysis and Bayesian skyline plots to infer historical demography. J. Hered. 2015, 106, 333–346. [Google Scholar] [CrossRef]
- Ho, P.T.; Kwan, Y.S.; Kim, B.; Won, Y.J. Postglacial range shift and demographic expansion of the marine intertidal snail Batillaria attramentaria. Ecol. Evol. 2015, 5, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Onsins, S.E.; Rozas, J. Statistical properties of new neutrality tests against population growth. Mol. Biol. Evol. 2002, 19, 2092–2100. [Google Scholar] [CrossRef] [PubMed]
- Sano, A.; Tachida, H. Gene genealogy and properties of test statistics of neutrality under population growth. Genetics 2005, 169, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R. Statistical tests of selective neutrality in the age of genomics. Heredity 2001, 86, 641–647. [Google Scholar] [CrossRef]
- Rogers, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 1992, 9, 552–569. [Google Scholar] [CrossRef]
- Slatkin, M.; Hudson, R.R. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 1991, 129, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Kivisild, T. Founder effect. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar] [CrossRef]
- Peterson, C.; Barber, R.; Skilleter, G. Chapter 2: Global warming and coastal ecosystem response: How to Northern and Southern hemispheres may differ in the Eastern Pacific Ocean. In Earth Systema Responses to Global Change; Academic Press: Cambridge, MA, USA, 1993. [Google Scholar]
- Clark, P.U.; Dyke, A.S.; Shakun, J.D.; Carlson, A.E.; Clark, J.; Wohlfarth, B.; Mitrovica, J.; Hostetler, S.; McCabe, A.M. The last glacial maximum. Science 2009, 325, 710–714. [Google Scholar] [CrossRef]
- Jenkins, T.L.; Castilho, R.; Stevens, J.R. Meta-analysis of northeast Atlantic marine taxa shows contrasting phylogeographic patterns following post-LGM expansions. PeerJ 2018, 6, e5684. [Google Scholar] [CrossRef]
- Boissin, E.; Neglia, V.; Baksay, S.; Micu, D.; Bat, L.; Topaloglu, B.; Todorova, V.; Panayotova, M.; Kruschel, C.; Milchakova, N.; et al. Chaotic genetic structure and past demographic expansion of the invasive gastropod Tritia neritea in its native range, the Mediterranean Sea. Sci. Rep. 2020, 10, 21624. [Google Scholar] [CrossRef]
- Zelada-Mázmela, E.; Reyes-Flores, L.E.; Sánchez-Velásquez, J.J.; Ingar, C.; Santos-Rojas, L.E. Population structure and demographic history of the gastropod Thaisella chocolata (Duclos, 1832) from the Southeast Pacific inferred from mitochondrial DNA analyses. Ecol. Evol. 2022, 12, e9276. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Superfamily | Taxon | GenBank Number | Length pb | CDS Length pb | 3 CDS Length pb | Source |
|---|---|---|---|---|---|---|
| Fissurelloidea | M. crenulata—PCA | OR911353 | 16,788 | 11,294 | 1488 | This study |
| Fissurelloidea | M. crenulata—PEU | PQ152234 | 16,787 | 11,118 | 1449 | This study |
| Fissurelloidea | M. crenulata—CAL (ATP8) | PQ152231 | - | - | 1488 | This study |
| Fissurelloidea | M. crenulata—CAL (NAD6) | PQ152232 | - | - | 1488 | This study |
| Fissurelloidea | M. crenulata—CAL (COX3) | PQ152233 | - | - | 1488 | This study |
| Fissurelloidea | Diodora graeca | KT207825 | 17,209 | 11,535 | 1498 | [18] |
| Fissurelloidea | Fissurella volcano | NC_016953 | 17,575 | 11,412 | 1434 | [24] |
| Haliotoidea | Haliotis iris | NC_031361 | 17,131 | 11,262 | 1458 | [25] |
| Haliotoidea | Haliotis rubra | NC_005940 | 16,907 | 11,253 | 1455 | [26] |
| Haliotoidea | Haliotis rufescens | NC_036928 | 16,646 | 11,253 | 1452 | [27] |
| Haliotoidea | Haliotis discus hannai | EU595789 | 15,784 | 11,055 | 1287 | [28] |
| Lepetodriloidea | Lepetodrilus schrolli | KR297250 | 15,579 | 11,406 | 1518 | [18] |
| Locality | Genetic Diversity | Neutrality Analyses | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | H | S | h ± S.D. | π ± S.D. | θ ± S.D. | DT | p-Value | Fs | p-Value | R2 | p-Value | |
| CAL | 19 | 10 | 13 | 0.906 ± 0.045 | 0.0027 ± 0.0002 | 3.130 ± 1.697 | −0.589 | 0.302 | −2.792 | 0.101 | 0.105 | 0.190 |
| ENS | 25 | 13 | 24 | 0.903 ± 0.038 | 0.0028 ± 0.0005 | 3.292 ± 1.751 | −1.778 | 0.019 | −4.662 | 0.050 | 0.078 | 0.057 |
| SQT | 16 | 11 | 12 | 0.925 ± 0.050 | 0.0028 ± 0.0004 | 3.300 ± 1.790 | −0.343 | 0.407 | −4.763 | 0.017 | 0.121 | 0.298 |
| SJO | 14 | 11 | 14 | 0.956 ± 0.045 | 0.0030 ± 0.0004 | 3.570 ± 1.928 | −0.775 | 0.242 | −5.444 | 0.011 | 0.092 | 0.027 |
| PCA | 15 | 10 | 18 | 0.924 ± 0.053 | 0.0033 ± 0.0005 | 3.840 ± 1.044 | −1.252 | 0.097 | −3.115 | 0.078 | 0.081 | 0.010 |
| PUE | 21 | 13 | 16 | 0.914 ± 0.049 | 0.0027 ± 0.0003 | 3.179 ± 1.712 | −1.054 | 0.145 | −6.002 | 0.013 | 0.081 | 0.039 |
| BAS | 8 | 6 | 10 | 0.893 ± 0.111 | 0.0027 ± 0.0006 | 3.150 ± 1.821 | −0.916 | 0.202 | −1.500 | 0.143 | 0.126 | 0.063 |
| IGP | 7 | 3 | 2 | 0.524 ± 0.209 | 0.0005 ± 0.0002 | 0.571 ± 0.521 | −1.237 | 0.000 | −0.922 | 0.173 | 0.226 | 0.560 |
| Global | 125 | 46 | 48 | 0.915 ± 0.015 | 0.0028 ± 0.0001 | 3.004 ± 1.861 | −1.944 | 0.004 | −33.240 | 0.000 | 0.034 | 0.003 |
| CAL | ENS | SQT | SJO | PCA | PEU | BAS | IGP | |
|---|---|---|---|---|---|---|---|---|
| CAL | 0.279 | 0.495 | 0.855 | 0.693 | 0.603 | 0.018 | 0.045 | |
| ENS | 0.009 | 0.018 | 0.162 | 0.396 | 0.612 | 0.153 | 0.072 | |
| SQT | −0.001 | 0.113 | 0.414 | 0.198 | 0.054 | 0.001 | 0.018 | |
| SJO | −0.035 | 0.024 | −0.009 | 0.459 | 0.405 | 0.018 | 0.027 | |
| PCA | −0.024 | 0.000 | 0.036 | −0.007 | 0.315 | 0.001 | 0.261 | |
| PEU | −0.015 | −0.015 | 0.073 | −0.004 | 0.004 | 0.135 | 0.000 | |
| BAS | 0.134 | 0.040 | 0.235 | 0.101 | 0.134 | 0.045 | 0.000 | |
| IGP | 0.113 | 0.079 | 0.205 | 0.124 | 0.023 | 0.142 | 0.356 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonett-Calzada, B.; Valenzuela-Quiñonez, F.; Del Río-Portilla, M.A.; Bayona-Vásquez, N.J.; Vargas-Peralta, C.E.; Hyde, J.R.; Lafarga-De la Cruz, F. Genetic Insights into the Giant Keyhole Limpet (Megathura crenulata), an Eastern Pacific Coastal Endemic: Complete Mitogenome, Phylogenetics, Phylogeography, and Historical Demography. Genes 2024, 15, 1303. https://doi.org/10.3390/genes15101303
Bonett-Calzada B, Valenzuela-Quiñonez F, Del Río-Portilla MA, Bayona-Vásquez NJ, Vargas-Peralta CE, Hyde JR, Lafarga-De la Cruz F. Genetic Insights into the Giant Keyhole Limpet (Megathura crenulata), an Eastern Pacific Coastal Endemic: Complete Mitogenome, Phylogenetics, Phylogeography, and Historical Demography. Genes. 2024; 15(10):1303. https://doi.org/10.3390/genes15101303
Chicago/Turabian StyleBonett-Calzada, Brenda, Fausto Valenzuela-Quiñonez, Miguel A. Del Río-Portilla, Natalia J. Bayona-Vásquez, Carmen E. Vargas-Peralta, John R. Hyde, and Fabiola Lafarga-De la Cruz. 2024. "Genetic Insights into the Giant Keyhole Limpet (Megathura crenulata), an Eastern Pacific Coastal Endemic: Complete Mitogenome, Phylogenetics, Phylogeography, and Historical Demography" Genes 15, no. 10: 1303. https://doi.org/10.3390/genes15101303