
Germplasm Resources Evaluation of Cultured Largemouth Bass (Micropterus salmoides) in China Based on Whole Genome Resequencing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Sequencing
2.3. Genotyping
2.4. Genetic Diversity and Linkage Disequilibrium Analysis
2.5. Population Structure Analysis
2.6. Gene Flow Analysis
3. Results
3.1. Summary of Sequencing and SNP Calling
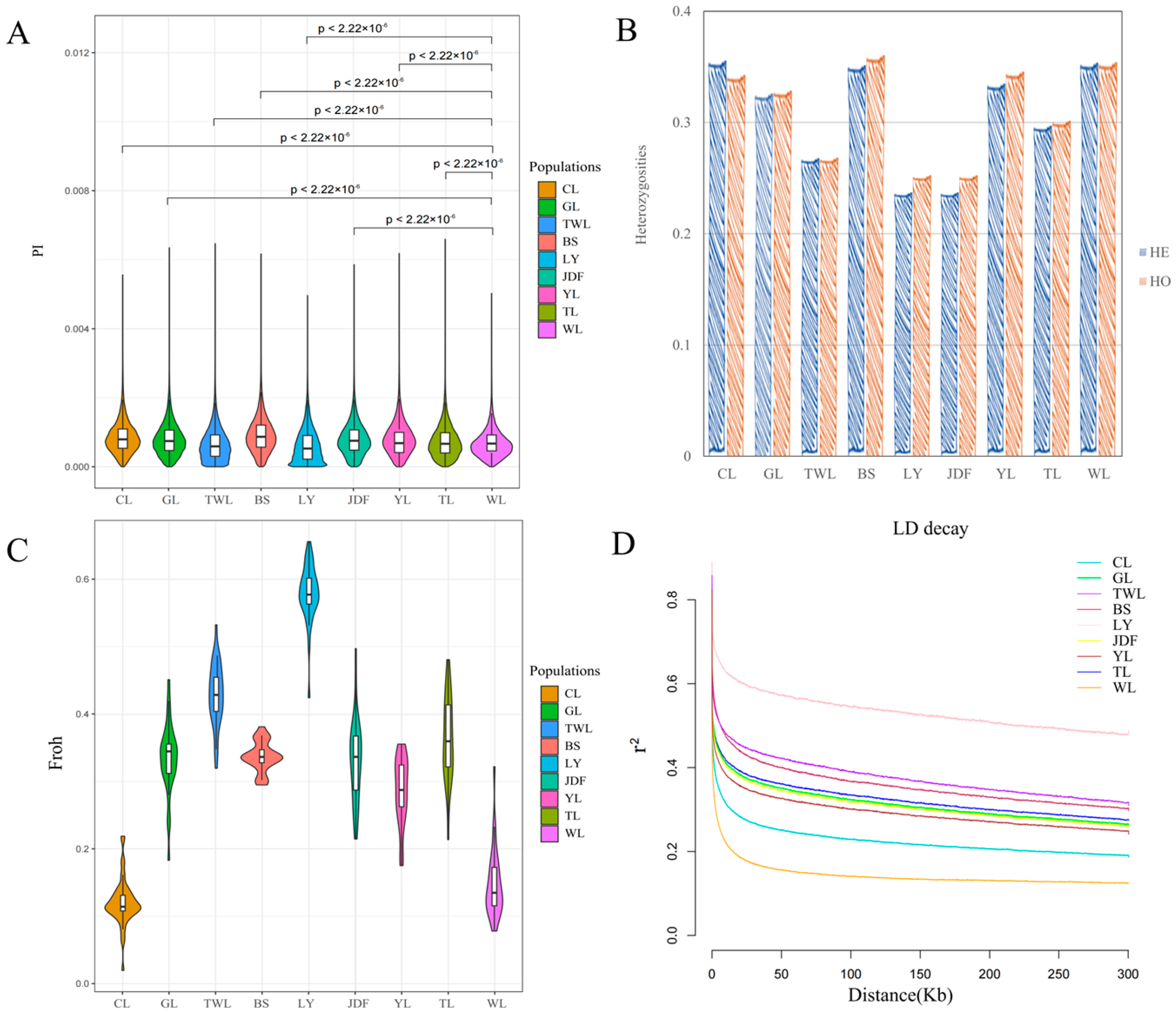
3.2. Results of Genetic Diversity and LD Analysis
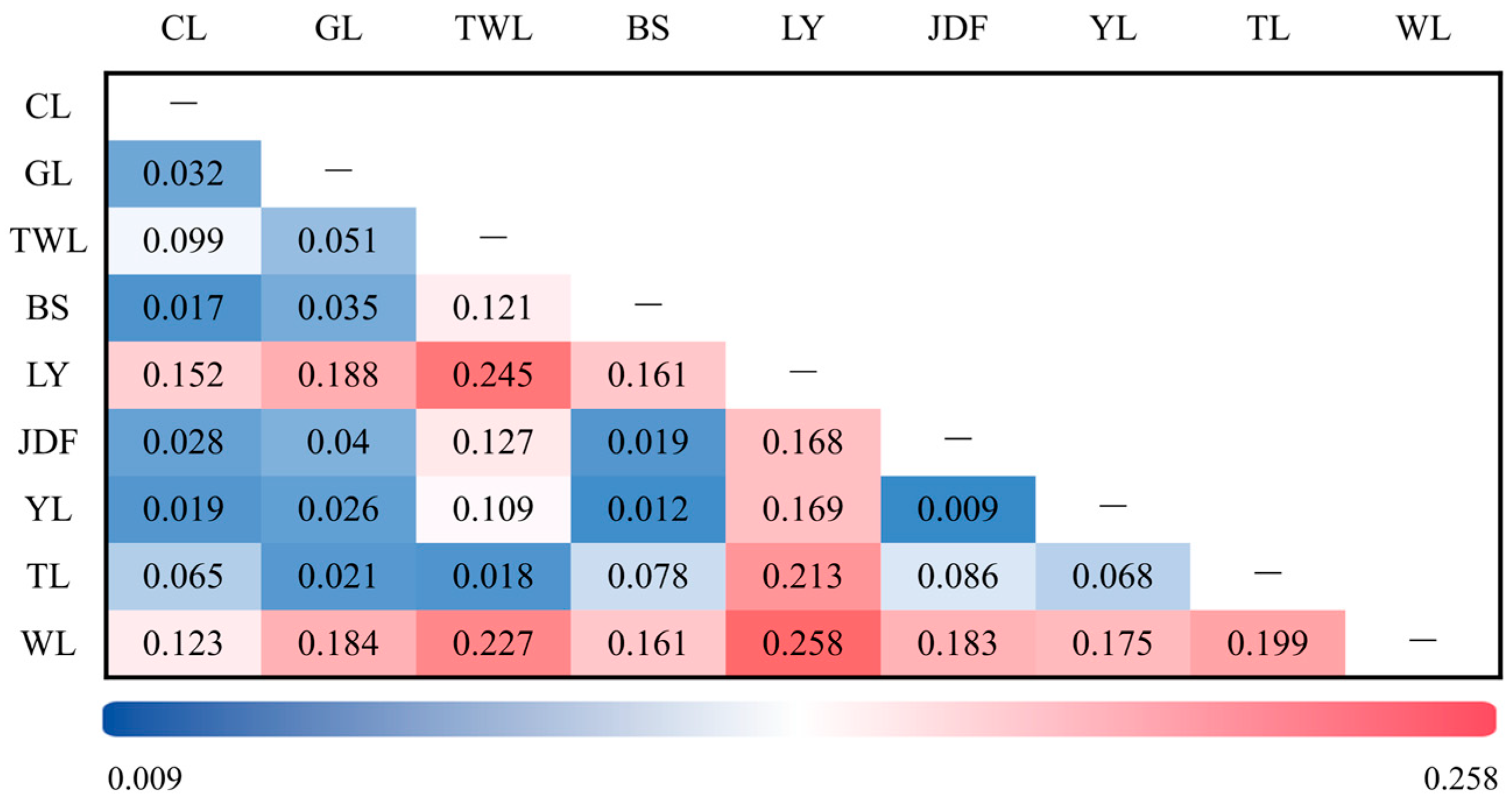
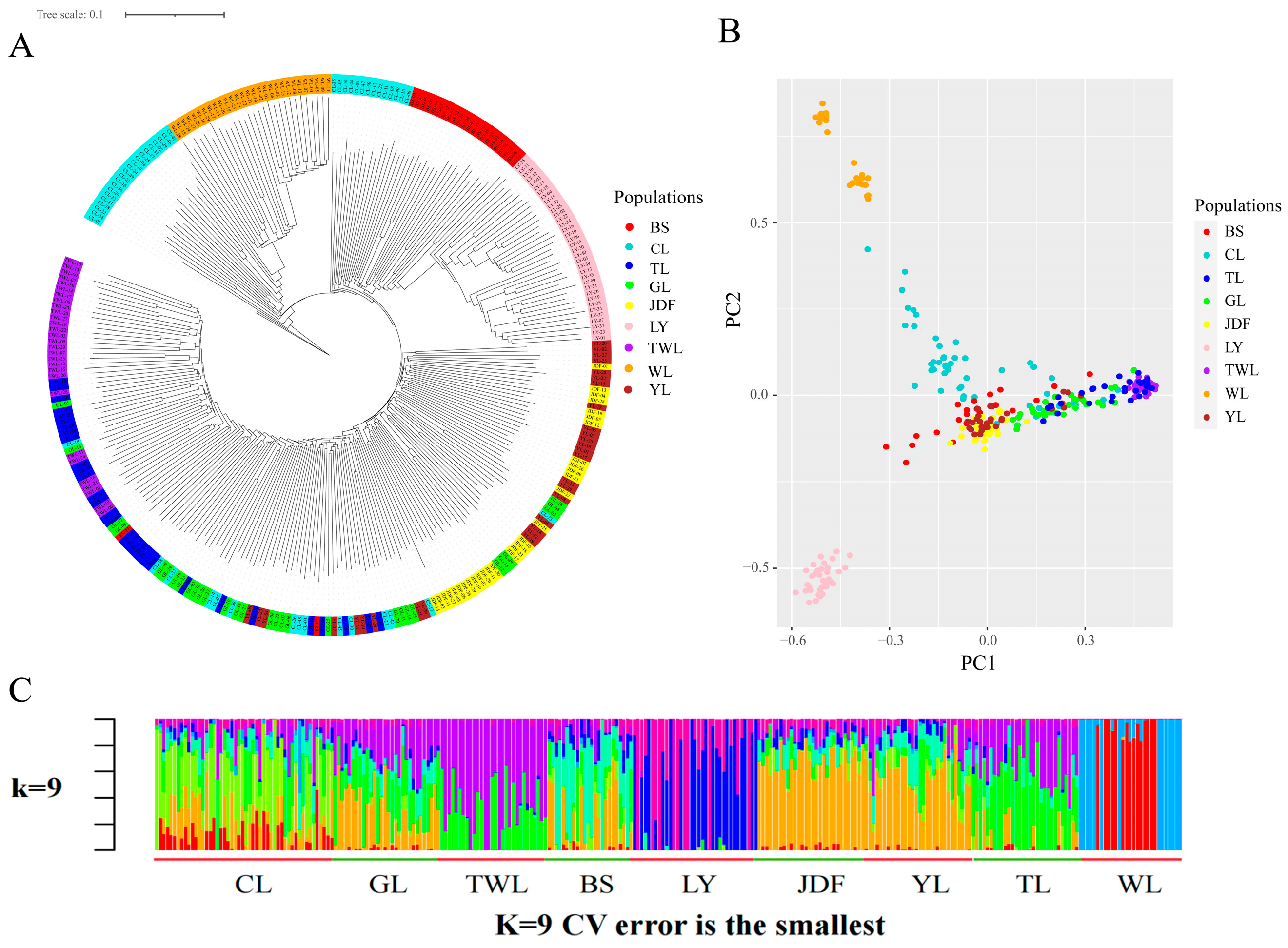
3.3. Results of Genetic Differentiation
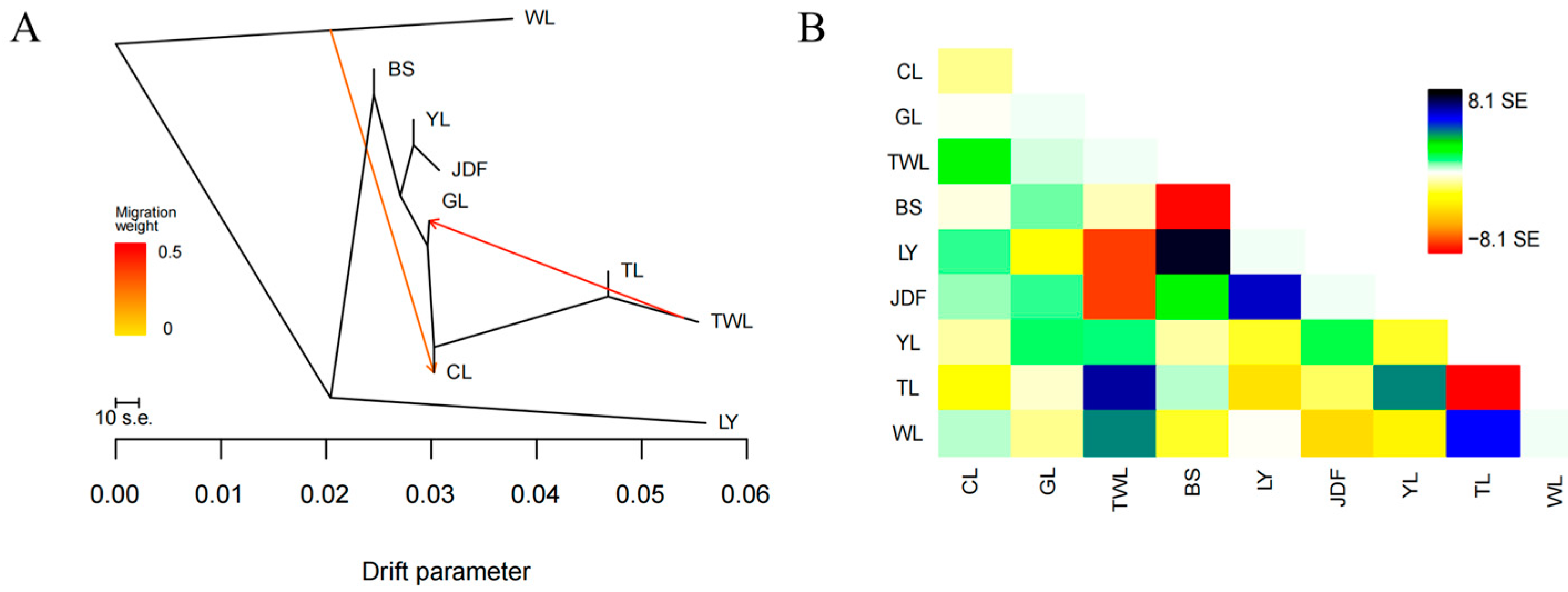
3.4. Gene Flow
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bai, J.; Lutz-Carrillo, D.J.; Quan, Y.; Liang, S. Taxonomic status and genetic diversity of cultured largemouth bass Micropterus salmoides in China. Aquaculture 2008, 278, 27–30. [Google Scholar] [CrossRef]
- Ribeiro, F.; Elvira, B.; Collares-Pereira, M.J.; Moyle, P.B. Life-history traits of non-native fishes in Iberian watersheds across several invasion stages: A first approach. Biol. Invasions 2008, 10, 89–102. [Google Scholar] [CrossRef]
- Beamish, C.A.; Booth, A.J.; Deacon, N. Age, growth and reproduction of largemouth bass, Micropterus salmoides, in Lake Manyame, Zimbabwe. Afr. Zool. 2005, 40, 63–69. [Google Scholar] [CrossRef]
- Wang, D.; Yao, H.; Li, Y.H.; Xu, Y.J.; Ma, X.F.; Wang, H.P. Global diversity and genetic landscape of natural populations and hatchery stocks of largemouth bass Micropterus salmoides across American and Asian regions. Sci. Rep. 2019, 9, 16697. [Google Scholar] [CrossRef]
- Yu, P.; Chen, H.; Liu, M.; Zhong, H.; Wang, X.; Wu, Y.; Sun, Y.; Wu, C.; Wang, S.; Zhao, C.; et al. Current status and application of largemouth bass (Micropterus salmoides) germplasm resources. Reprod. Breed. 2024, 4, 73–82. [Google Scholar] [CrossRef]
- Bailey, R.; Hubbs, C. The Black Basses (Micropterus) of Florida, with Description of a New Species; University of Michigan Occasional Papers of the Museum of Zoology: Ann Arbor, MI, USA, 1949. [Google Scholar]
- Bai, J.J.; Li, S.J. Aquaculture in China: Success Stories and Modern Trends; Gui, J.F., Tang, Q.S., Li, Z.J., Liu, L.S., De Silva, S.S., Eds.; John Wiley & Sons Ltd: New York, NY, USA, 2018; pp. 421–429. [Google Scholar]
- Bureau of Fisheries. China Fishery Statistical Yearbook; Bureau of Fisheries; Chinese Ministry of Agriculture: Beijing, China, 2023; Available online: https://deepblue.lib.umich.edu/handle/2027.42/56954 (accessed on 25 September 2024). (In Chinese)
- Zhu, X.; Qian, Q.; Wu, C.; Zhu, Y.; Gao, X.; Jiang, Q.; Wang, J.; Liu, G.; Zhang, X. Pathogenicity of Aeromonas veronii Causing Mass Mortality of Largemouth Bass (Micropterus salmoides) and Its Induced Host Immune Response. Microorganisms 2022, 10, 2198. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.N.; Zhong, Y.; Luo, M.J.; Zheng, G.; Huang, J.L.; Wang, G.P.; Geng, Y.; Qian, X.Q. Largemouth bass ranavirus: Current status and research progression. Aquacult. Rep. 2023, 32, 101706. [Google Scholar] [CrossRef]
- Philipp, D.P.; Whitt, G.S. Survival and growth of northern, florida, and reciprocal f1-hybrid largemouth bass in Central Illinois. Trans. Am. Fish. Soc. 1991, 120, 58–64. [Google Scholar] [CrossRef]
- Williamson, J.H.; Carmichael, G.J. An aquacultural evaluation of florida, northern, and hybrid largemouth bass, Micropterus salmoides. Aquaculture 1990, 85, 247–257. [Google Scholar] [CrossRef]
- Zolczynski, S.J.; Davies, W.D. Growth-characteristics of northern and florida subspecies of largemouth bass and their hybrid, and a comparison of catchability between subspecies. Trans. Am. Fish. Soc. 1976, 105, 240–243. [Google Scholar] [CrossRef]
- Huang, R.; Zhong, C.; Zhang, D.; Qiang, J.; Tao, Y.; Li, Y.; Dong, Y.; Lu, S.; Xu, P. Comparative study on growth performance, muscle nutrients, intestinal morphology and structure and microflora composition of different largemouth bass (Micropterus salmoides) populations. Chin. J. Anim. Nutr. 2024, 36, 1158–1172. [Google Scholar]
- Zhou, J.; Li, S.; Jiang, P.; Sun, X.; Han, L.; Bai, J. Comparison analysis of genetic diversity and growth traits among “Youlu No.1”and their reciprocal hybrids of northern Micropterus salmoides. Marine Fisheries 2020, 42, 324–331. (In Chinese) [Google Scholar]
- Neal, J.W.; Neal, D.M.; Noble, R.L.; McGee, M.V. Artificial propagation and induction of triploidy in largemouth bass Micropterus salmoides and ploidy discrimination using erythrocyte length. J. World Aquacult. Soc. 2004, 35, 46–54. [Google Scholar] [CrossRef]
- Xu, L.; Bai, J.; Li, S.; Fan, J. Analysis of growth-related genotypes in “Youlu No.1” largemouth bass (Micropterus salmoides). Marine Fisheries 2014, 36, 24–28. (In Chinese) [Google Scholar]
- Li, S.J.; Bai, J.J.; Han, L.Q.; Sun, J.G. Breeding example of “Youlu No. 3” largemouth bass (Micropterus salmoides). Sci. Fish Farm 2018, 9, 44–45. (In Chinese) [Google Scholar]
- Shen, L.Y. A new result in the selection and breeding of the new variety of “Wanlu No.1” largemouth bass (Micropterus salmoides). Fish. Sci. Technol. Inf. 2022, 46, 174. [Google Scholar]
- A New Strain of “Jia Defeng” Largemouth Bass (Micropterus salmoides) Has Been Bred to Produce a Good Harvest. Available online: https://www.prfri.ac.cn/info/1143/6230.htm (accessed on 25 September 2024). (In Chinese).
- Zhu, X.; Du, H.; Guangming, Z.; Zhongyao, J.; Sai, C.; Zian, C. Genetic diversity analysis of largemouth bass (Micropterus salmoides) cultured in ponds. J. Dalian Fish. Coll. 2006, 21, 341–345. (In Chinese) [Google Scholar]
- Suxian, L.; Junjie, B.A.I.; Xing, Y.E.; Haihua, L.A.O.; Qing, J. RAPD analysis of genetic diversity for cultured largemouth bass Micropterus salmoides. J. Dalian Fish. Coll. 2007, 22, 260–263. (In Chinese) [Google Scholar]
- Zhang, D.; Qiang, J.; Fu, J.; Xu, G.; Zhou, G.; Dong, Z.; Tao, Y.; Lu, S.; Lu, J.; Xu, P. Genetic analysis of five stocks of largemouth bass (Micropterus salmoides) with microsatellite and mitochondrial D-loop sequences. J. Fish. Sci. China 2022, 29, 1277–1289. (In Chinese) [Google Scholar]
- Zhang, D.; Dong, S.; Bai, J.; Miao, J.; Hu, Y.; Li, S.; Miao, J.; Zhang, X.; Ma, D. Sequence analysis of mtDNA COI region in northern and Florida subspecies of largemouth bass Micropterus salmoides. J. Dalian Ocean. Univ. 2014, 29, 212–216. (In Chinese) [Google Scholar]
- Liang, S.; Sun, X.; Bai, J.; Gao, J. Genetic analysis for cultured largemouth bass (Micropterus salmoides) in China with microsatellites. Acta Hydrobiol. Sin. 2008, 32, 694–700. (In Chinese) [Google Scholar] [CrossRef]
- Du, J.; Li, S.; Shao, J.; Song, H.; Jiang, P.; Lei, C.; Bai, J.; Han, L. Genetic diversity analysis and development of molecular markers for the identification of largemouth bass (Micropterus salmoides L.) based on whole-genome re-sequencing. Front. Genet. 2022, 13, 936610. [Google Scholar] [CrossRef]
- Sun, C.F.; Zhang, X.H.; Dong, J.J.; You, X.X.; Tian, Y.Y.; Gao, F.Y.; Zhang, H.T.; Shi, Q.; Ye, X. Whole-genome resequencing reveals recent signatures of selection in five populations of largemouth bass ( Micropterus salmoides). Zool. Res. 2023, 44, 78–89. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2017, 7, gix120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data, P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Abraham, G.; Qiu, Y.; Inouye, M. FlashPCA2: Principal component analysis of Biobank-scale genotype datasets. Bioinformatics 2017, 33, 2776–2778. [Google Scholar] [CrossRef] [PubMed]
- Ginestet, C. ggplot2: Elegant Graphics for Data Analysis. J. R. Stat. Soc. A. Stat. Soc. 2011, 174, 245–246. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. The interpretation of population-structure by F-statistics with special regard to systems of mating. Evolution 1965, 19, 395–420. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Pritchard, J.K. Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef] [PubMed]
- Fitak, R.R. OptM: Estimating the optimal number of migration edges on population trees using Treemix. Biol. Methods Protoc. 2021, 6, bpab017. [Google Scholar] [CrossRef]
- Pei, C.; Song, H.L.; Zhu, L.; Qiao, D.; Yan, Y.; Li, L.; Zhao, X.L.; Zhang, J.; Jiang, X.Y.; Kong, X.H. Identification of Aeromonas veronii isolated from largemouth bass Micropterus salmoides and histopathological analysis. Aquaculture 2021, 540, 736707. [Google Scholar] [CrossRef]
- Liu, Q.H.; Lin, H.D.; Chen, J.; Ma, J.K.; Liu, R.Q.; Ding, S.X. Genetic variation and population genetic structure of the large yellow croaker (Larimichthys crocea) based on genome-wide single nucleotide polymorphisms in farmed and wild populations. Fish. Res. 2020, 232, 105718. [Google Scholar] [CrossRef]
- Tang, H.P.; Mao, S.Q.; Xu, X.Y.; Li, J.L.; Shen, Y.B. Genetic diversity analysis of different geographic populations of black carp (Mylopharyngodon piceus) based on whole genome SNP markers. Aquaculture 2024, 582, 740542. [Google Scholar] [CrossRef]
- Buckler, E.S.; Thornsberry, J.M. Plant molecular diversity and applications to genomics. Curr. Opin. Plant Biol. 2002, 5, 107–111. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, S.; Ren, H.; Tang, J.; Gao, J.; Li, X.; Fu, Z.; He, Z. Genetic diversity and population structure of the endangered Japanese sea cucumber (Apostichopus japonicus) in natural seas of northern China. Aquacult. Rep. 2023, 30, 101595. [Google Scholar] [CrossRef]
- Zhou, X.; Yi, S.; Zhao, W.; Zhou, Q.; Shen, J.; Li, D.; Huo, B.; Tang, R. Genetic diversity and population differentiation of kashgarian loach (Triplophysa yarkandensis) in Xinjiang Tarim River Basin. Biology 2021, 10, 734. [Google Scholar] [CrossRef]
- Su, S.; Zhang, L.; Li, H.; Gao, C.; He, X.; Tian, C.; Li, J.; Wang, M.; Tang, Y. Genetic diversity and structure analyses of large-mouth bass (Micropterus salmoides) original and cultured populations based on microsatellite markers. J. Zhejiang Uni. (Agric. Life Sci.) 2020, 46, 687–698. (In Chinese) [Google Scholar]
- Arcos-Burgos, M.; Muenke, M. Genetics of population isolates. Clin. Genet. 2002, 61, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, L.; Palotie, A.; Lange, K. Use of population isolates for mapping complex traits. Nat. Rev. Genet. 2000, 1, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.M.; Mulle, J.G.; Dodd, A.F.; Pulver, A.E.; Wooding, S.; Warren, S.T. Signatures of founder effects, admixture, and selection in the Ashkenazi Jewish population. Proc. Natl. Acad. Sci. USA 2010, 107, 16222–16227. [Google Scholar] [CrossRef]
- Chakraborty, R.; Weiss, K.M. Admixture as a tool for finding linked genes and detecting that difference from allelic association between loci. Proc. Natl. Acad. Sci. USA 1988, 85, 9119–9123. [Google Scholar] [CrossRef]
- Wilson, J.F.; Goldstein, D.B. Consistent long-range linkage disequilibrium generated by admixture in a Bantu-Semitic hybrid population. Am. J. Hum. Genet. 2000, 67, 926–935. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, W.; Jian, J.; Niu, B.; Zhang, X.; Yu, J.; Xu, X. Germplasm Resources Evaluation of Cultured Largemouth Bass (Micropterus salmoides) in China Based on Whole Genome Resequencing. Genes 2024, 15, 1307. https://doi.org/10.3390/genes15101307
Guan W, Jian J, Niu B, Zhang X, Yu J, Xu X. Germplasm Resources Evaluation of Cultured Largemouth Bass (Micropterus salmoides) in China Based on Whole Genome Resequencing. Genes. 2024; 15(10):1307. https://doi.org/10.3390/genes15101307
Chicago/Turabian StyleGuan, Wenzhi, Jieliang Jian, Baolong Niu, Xinhui Zhang, Jiongying Yu, and Xiaojun Xu. 2024. "Germplasm Resources Evaluation of Cultured Largemouth Bass (Micropterus salmoides) in China Based on Whole Genome Resequencing" Genes 15, no. 10: 1307. https://doi.org/10.3390/genes15101307
APA StyleGuan, W., Jian, J., Niu, B., Zhang, X., Yu, J., & Xu, X. (2024). Germplasm Resources Evaluation of Cultured Largemouth Bass (Micropterus salmoides) in China Based on Whole Genome Resequencing. Genes, 15(10), 1307. https://doi.org/10.3390/genes15101307