

Whole Exome Sequencing and Panel-Based Analysis in 176 Spanish Children with Neurodevelopmental Disorders: Focus on Autism Spectrum Disorder and/or Intellectual Disability/Global Developmental Delay
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants and Clinical Information
2.2. Ethical Statement and Consent
2.3. Data Collection
2.4. Whole Exome Sequencing
2.5. Sanger DNA Sequencing
2.6. Variant Inclusion and Classification
2.7. Statistical Analysis
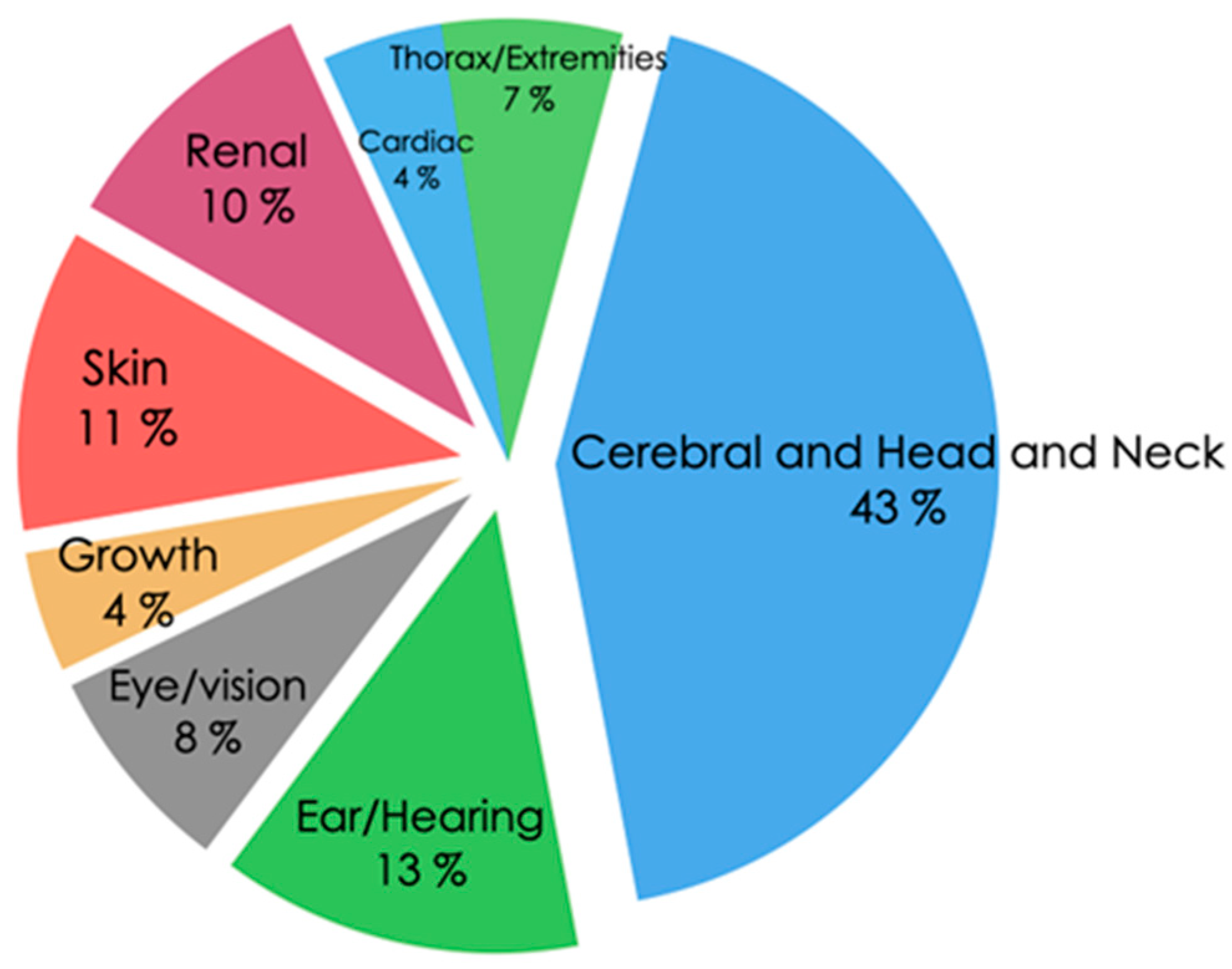
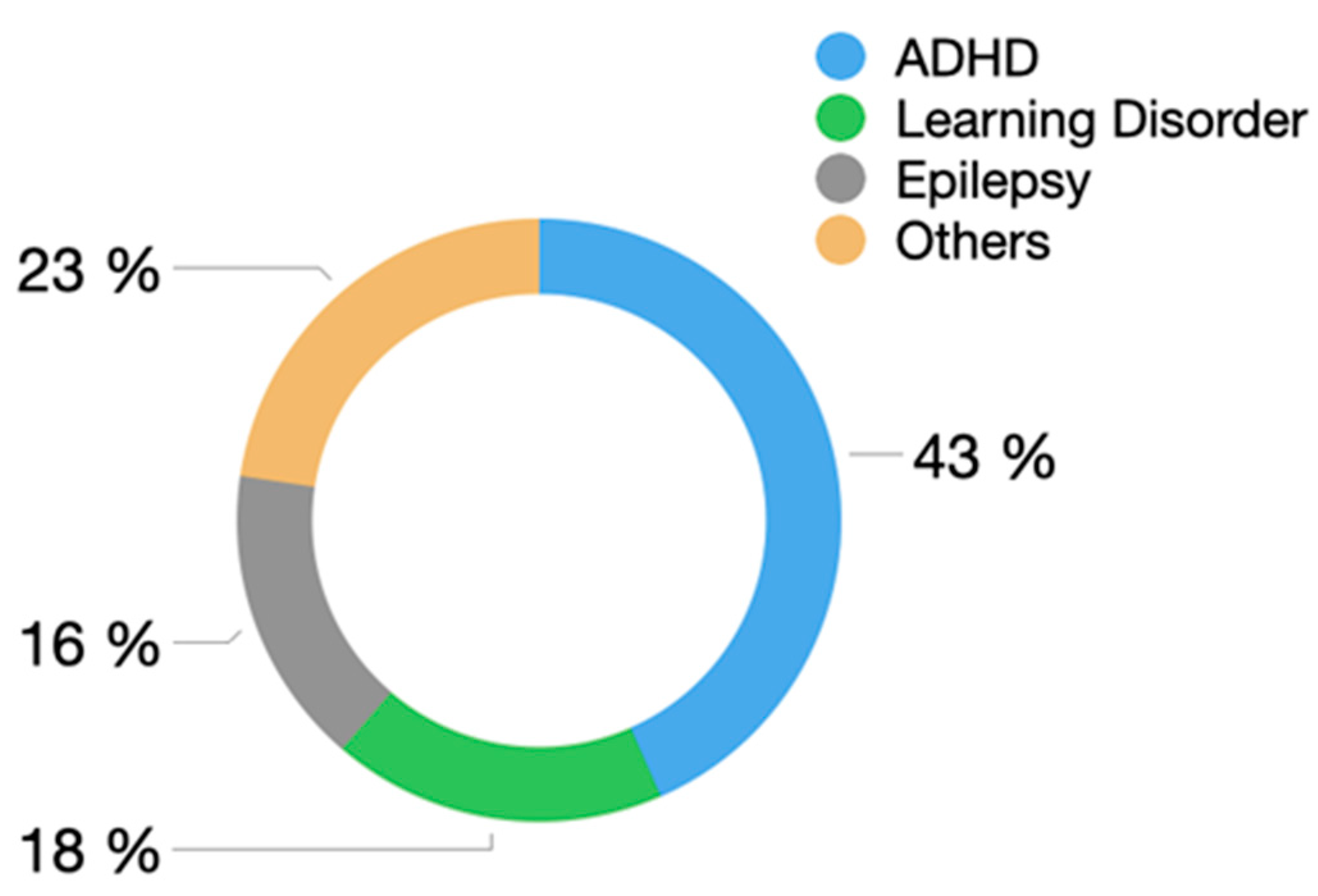
3. Results
4. Discussion
Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Arlington, VA, USA, 2013. [Google Scholar]
- Van Kamebeek, C.D.M. Evaluation of the Child with Develpmental Impairments. Contin. Child Neurol. 2018, 24, 228–247. [Google Scholar]
- Hirota, T.; King, B.H. Autism Spectrum Disorder. JAMA 2023, 329, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Totsika, V.; Liew, A.; Absoud, M.; Adnams, C.; Emerson, E. Mental health problems in children with intellectual disability. Lancet Child Adolesc. Health 2022, 6, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Taşkıran, E.Z.; Karaosmanoğlu, B.; Koşukcu, C.; Ürel-Demir, G.; Akgün-Doğan, Ö.; Şimşek-Kiper, P.Ö.; Alikaşifoğlu, M.; Boduroğlu, K.; Utine, G.E. Diagnostic yield of whole-exome sequencing in non-syndromic intellectual disability. J. Intellect. Disabil. Res. 2021, 65, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Mora, M.I.; Sánchez, A.; Rodríguez-Revenga, L.; Corominas, J.; Rabionet, R.; Puig, S.; Madrigal, I. Diagnostic yield of next-generation sequencing in 87 families with neurodevelopmental disorders. Orphanet J. Rare Dis. 2022, 17, 60. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Márquez-Caraveo, M.E.; Rodríguez-Valentín, R.; Pérez-Barrón, V.; Vázquez-Salas, R.A.; Sánchez-Ferrer, J.C.; De Castro, F.; Allen-Leigh, B.; Lazcano-Ponce, E. Children and adolescents with neurodevelopmental disorders show cognitive heterogeneity and require a person-centered approach. Sci. Rep. 2021, 11, 18463. [Google Scholar] [CrossRef]
- Stefanski, A.; Calle-López, Y.; Leu, C.; Pérez-Palma, E.; Pestana-Knight, E.; Lal, D. Clinical sequencing yield in epilepsy, autism spectrum disorder, and intellectual disability: A systematic review and meta-analysis. Epilepsia 2020, 62, 143–151. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, S.H.; Kim, B.; Lee, J.S.; Kim, H.D.; Choi, J.R.; Lee, S.-T.; Kang, H.-C.; Kim, S.H.; Kim, B.; Lee, J.S.; et al. Proband-Only Clinical Exome Sequencing for Neurodevelopmental Disabilities. Pediatr. Neurol. 2019, 99, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Tammimies, K.; Marshall, C.R.; Walker, S.; Kaur, G.; Thiruvahindrapuram, B.; Lionel, A.C. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with Autism spectrum disorder. JAMA 2015, 314, 895. [Google Scholar] [CrossRef]
- Xiao, B.; Qiu, W.; Ji, X.; Liu, X.; Huang, Z.; Liu, H.; Fan, Y.; Xu, Y.; Liu, Y.; Yie, H.; et al. Marked yield of re-evaluating phenotype and exome/target sequencing data in 33 individuals with intellectual disabilities. Am. J. Med. Genet. Part A 2017, 176, 107–115. [Google Scholar] [CrossRef]
- Srivastava, S.; Love-Nichols, J.A.; Dies, K.A.; Ledbetter, D.H.; Martin, C.L.; Chung, W.K.; Firth, H.V.; Frazier, T.; Hansen, R.L.; Prock, L.; et al. NDD Exome Scoping Review Work Group. Correction: Meta-analysis and multidisciplinary consensus statement: Exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet. Med. 2020, 22, 1731–1732. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Valentino, F.; Bruno, L.P.; Doddato, G.; Giliberti, A.; Tita, R.; Resciniti, S.; Fallerini, C.; Bruttini, M.; Rizzo, C.L.; Mencarelli, M.A.; et al. Exome Sequencing in 200 Intellectual Disability/Autistic Patients: New Candidates and Atypical Presentations. Brain Sci. 2021, 11, 936. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Arteche-López, A.; Rodríguez, M.J.G.; Calvin, M.T.S.; Quesada-Espinosa, J.F.; Rosales, J.M.L.; Milla, C.P.; Gómez-Manjón, I.; Mayoral, I.H.; de la Fuente, R.P.; de Bustamante, A.D.; et al. Towards a Change in the Diagnostic Algorithm of Autism Spectrum Disorders: Evidence Supporting Whole Exome Sequencing as a First-Tier Test. Genes 2021, 12, 560. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rossi, M.; El-Khechen, D.; Black, M.H.; Hagman, K.D.F.; Tang, S.; Powis, Z. Outcomes of Diagnostic Exome Sequencing in Patients with Diagnosed or Suspected Autism Spectrum Disorders. Pediatr. Neurol. 2017, 70, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Corominas, R.; Lin, G.N. De novo Mutations from Whole Exome Sequencing in Neurodevelopmental and Psychiatric Disorders: From Discovery to Application. Front. Genet. 2019, 10, 258. [Google Scholar] [CrossRef]
- Du, X.; Gao, X.; Liu, X.; Shen, L.; Wang, K.; Fan, Y.; Sun, Y.; Luo, X.; Liu, H.; Wang, L.; et al. Genetic Diagnostic Evaluation of Trio-Based Whole Exome Sequencing among Children with Diagnosed or Suspected Autism Spectrum Disorder. Front. Genet. 2018, 9, 594. [Google Scholar] [CrossRef]
- Devi, R.U.; Kumar, J.T.; Jan, S.M.S.; Chandrasekaran, A.; Amboiram, P.; Koshy, T.; Balakrishnan, U. Utility of clinical exome sequencing in the evaluation of neonates with suspected genetic condition—An observational study from tertiary neonatal care unit in South India. Eur. J. Med Genet. 2021, 64, 104247. [Google Scholar] [CrossRef] [PubMed]
- Vinci, M.; Treccarichi, S.; Rando, R.G.; Musumeci, A.; Todaro, V.; Federico, C.; Saccone, S.; Elia, M.; Calì, F. A de novo ARIH2 gene mutation was detected in a patient with autism spectrum disorders and intellectual disability. Sci. Rep. 2024, 14, 15848. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, K.; Bu, F.; Wu, Y.; Zhang, G.; Wang, X.; He, S.; Liu, M.-F.; Chen, R.; Yuan, H. Exploring noncoding variants in genetic diseases: From detection to functional insights. J. Genet. Genom. 2024, 51, 111–132. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, D. Wechsler Intelligence Scale for Children—Fifth Edition (WISC-V); Pearson: London, UK, 2014. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chand, R.P.; Vinit, W.; Vaidya, V.; Iyer, A.S.; Shelke, M.; Aggarwal, S.; Magar, S.; Danda, S.; Moirangthem, A.; Phadke, S.R.; et al. Proband only exome sequencing in 403 Indian children with neurodevelopmental disorders: Diagnostic yield, utility and challenges in a resource-limited setting. Eur. J. Med Genet. 2023, 66, 104730. [Google Scholar] [CrossRef] [PubMed]
- Wayhelova, M.; Vallova, V.; Broz, P.; Mikulasova, A.; Smetana, J.; Filkova, H.D.; Machackova, D.; Handzusova, K.; Gaillyova, R.; Kuglik, P. Exome sequencing improves the molecular diagnostics of paediatric unexplained neurodevelopmental disorders. Orphanet J. Rare Dis. 2024, 19, 41. [Google Scholar] [CrossRef]
- Savatt, J.M.; Myers, S.M. Genetic Testing in Neurodevelopmental Disorders. Front. Pediatr. 2021, 9, 526779. [Google Scholar] [CrossRef]
- Gonzalez-Mantilla, A.J.; Moreno-De-Luca, A.; Ledbetter, D.H.; Martin, C.L. A Cross-Disorder Method to Identify Novel Candidate Genes for Developmental Brain Disorders. JAMA Psychiatry 2016, 73, 275–283. [Google Scholar] [CrossRef]
- Wright, C.F.; Fitzgerald, T.W.; Jones, W.D.; Clayton, S.; McRae, J.F.; van Kogelenberg, M.; King, D.A.; Ambridge, K.; Barrett, D.M.; Bayzetinova, T.; et al. Genetic diagnosis of developmental disorders in the DDD study: A scalable analysis of genome-wide research data. Lancet 2015, 385, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Tsai, G.J.; Rañola, J.M.O.; Smith, C.; Garrett, L.T.; Bs, T.B.; Casadei, S.; Bowen, D.J.; Shirts, B.H. Outcomes of 92 patient-driven family studies for reclassification of variants of uncertain significance. Anesthesia Analg. 2018, 21, 1435–1442. [Google Scholar] [CrossRef]
- Thapar, A.; Rutter, M. Genetic Advances in Autism. J. Autism Dev. Disord. 2020, 51, 4321–4332. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.L.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584. [Google Scholar] [CrossRef]
- Myers, S.M.; Challman, T.D.; Bernier, R.; Bourgeron, T.; Chung, W.K.; Constantino, J.N.; Eichler, E.E.; Jacquemont, S.; Miller, D.T.; Mitchell, K.J.; et al. Insufficient Evidence for “Autism-Specific” Genes. Am. J. Hum. Genet. 2020, 106, 587–595. [Google Scholar] [CrossRef]
- Happé, F.; Ronald, A.; Plomin, R. Time to give up on a single explanation for autism. Nat. Neurosci. 2006, 9, 1218–1220. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Muzny, D.M.; Xia, F.; Niu, Z.; Person, R.; Ding, Y.; Ward, P.; Braxton, A.; Wang, M.; Buhay, C.; et al. Molecular Findings Among Patients Referred for Clinical Whole-Exome Sequencing. JAMA 2014, 312, 1870–1879. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.H.; Choi, S.H.; Yoo, H.W.; Kwak, M.J.; Park, K.H.; Kong, J.; Lee, Y.-J.; Nam, S.O.; Lee, B.L.; Chung, W.Y.; et al. Clinical use of whole exome sequencing in children with developmental delay/intellectual disability. Pediatr. Neonatol. 2024, 65, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M.; Ghazali, M.B.; Wahab, M.A.M.A.; Yusoff, N.M.; Mahsin, H.; Seng, C.E.; Khalid, I.A.; Rahman, M.N.G.; Yahaya, B.H. The BRCA1 and BRCA2 Genes in Early-Onset Breast Cancer Patients. Adv. Exp. Med. Biol. 2020, 1292, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women with Ovarian Cancer. JNCI J. Natl. Cancer Inst. 2015, 107, djv214. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Miller, D.T.; Lee, K.; Gordon, A.S.; Amendola, L.M.; Adelman, K.; Bale, S.J.; Chung, W.K.; Gollob, M.H.; Harrison, S.M.; Herman, G.E.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Anesthesia Analg. 2021, 23, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Gabis, L.V.; Gross, R.; Barbaro, J. Editorial: Personalized Precision Medicine in Autism Spectrum-Related Disorders. Front. Neurol. 2021, 12, 730852. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age (Mean; Range) | 6 Years (9 m–16 y) |
| Male sex, n (%) | 127 (72.1%) |
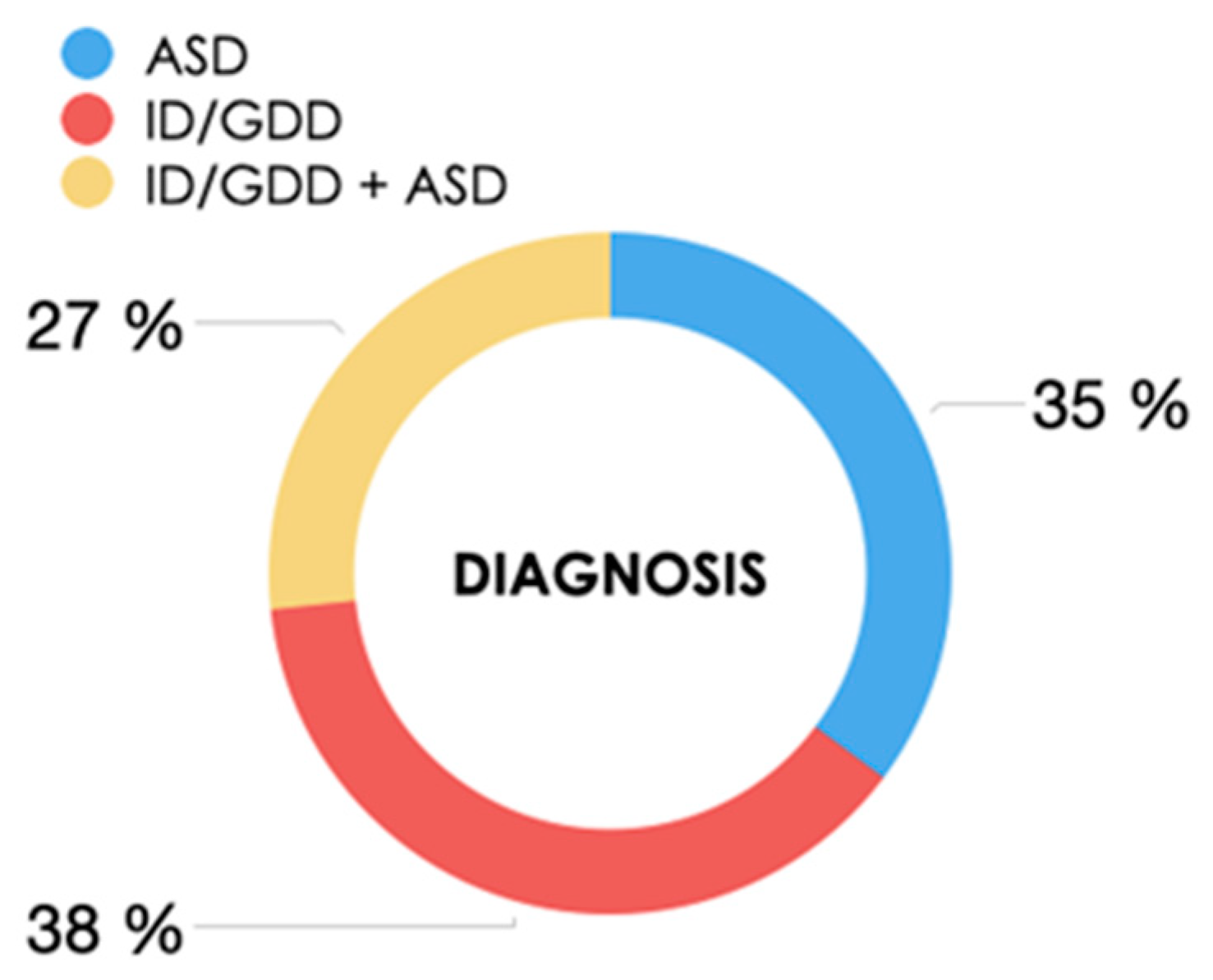
| Neurodevelopmental disorder, n (%): ID/GDD ASD ID/GDD + ASD | 67 (38.1%) 62 (35.2%) 47 (26.7%) |
| Ethnicity, n (%) Spanish Arab South American Other | 139 (79%) 16 (9%) 14 (8%) 7 (4%) |
| Mother’s age (mean) | 32 years |
| Father’s age (mean) | 35 years |
| Consanguinity, n (%) | 6 (3.4%) |
| Family history of ID and/or ASD, n (%) | 33 (18.8%) |
| Pregnancy, n (%) Spontaneous Assisted reproductive therapy | 164 (93.2%) 12 (6.8%) |
| Gestational age, n (%) Term deliveries Premature deliveries >32 weeks <32 weeks | 155 (88%) 21 (12%) 4 (18.7%) 17 (81.3%) |
| Delivery Eutocic/scheduled cesarean section Instrumental delivery/urgent cesarean section | 136 (77.2%) 40 (22.8%) |
| Diagnostic Yield by WES in Proband (n = 176) | |||
| ID/GDD group (n = 67) | ASD group (n = 62) | ID/GDD +ASD (n = 47) | Global |
| 20.8% | 3.2% | 12.7% | 12.5% |
| Diagnostic Yield after Studying Parental Segregation (n = 152) | |||
| ID/GDD group (n = 60) | ASD group (n = 53) | ID/GDD +ASD (n = 39) | Global |
| 26.6% | 1.8% | 23% | 17.1% |
| ID | Gene | Location | DNA Variants | Protein Variants | Zygosity | Inheritance | OMIM# | De Novo |
|---|---|---|---|---|---|---|---|---|
| P1 | HIVEP2 | 6q24.2 | c.737_740dup | p.(Val248IlefsTer11) | Het | AD | #616977 | de novo |
| P10 | CHAMP1 | 13q34 | c.1036G>T | Het | AD | #616579 | NA | |
| P28 | HUWE1 | Xp11.22 | c.12374A>G | p.(Glu4125Gly) | Hem | XL | #30959 | de novo |
| P40 | GATA3 | 10p14 | c.924+1G>A | Het | AD | #131320 | de novo | |
| P50 | KDM6B | 17p13.1 | c.1180C>G | p.(Pro394Ala) | Het | AD | #618505 | de novo |
| P51 | TLK2 | 17q23.2 | c.195_199del, | p.(Arg65SerfsTer2) | Het | AD | #618050 | NA |
| P60 | CNKSR2 | Xp22.12 | c.352C>T | p.(Arg118Ter) | Het | XL | #301008 | NA |
| P68 | MED13L | 12q24.21 | c.1280+1G>T | Het | AD | #616789 | de novo | |
| P69 | SHANK3 | 22q13.33 | c.3525delG | p.(Asp1176ThrfsTer4 | Het | AD | #606230 | de novo |
| P71 | CACNA1C | 12p13.33 | c.1113+5G>A | Het | AD | #601005 #618447 | de novo | |
| P80 | POGZ | 1q21.3 | c.2171dup | p.(Leu725SerfsTer19) | Het | AD | #614787 | NA |
| P89 | SOX5 | 12p12.1 | c.931+5G> | Het | AD | #616803 | de novo | |
| P107 | CUL3 | 2q36.2 | c.802delC | p.(Leu268SerfsTer5) | Het | AD | #619239 | de novo |
| P111 | NFIX | 19p13.13 | c.583+1G>A | Het | AD | #614753 | de novo | |
| P123 | CDKL5 | Xp22.13 | c.1675C>T | p.(Arg559Ter) | Het | XL | #300672 | NA |
| P128 | SPAST | 2p22.3 | c.1321+1G>A | Het | AD | #604277 | NA | |
| P135 | CIC | 19.13.2 | c.1331G>T | p.(Cys444Phe) | Het | AD | #617600 | de novo |
| P137 | DDX3X | Xp11.4 | c.1770-3_1770-2dup | Het | XL | #300958 | NA | |
| P144 | ASH1L | 1q22 | c.4025G>A | p.(Arg1342Gln) | Het | AD | #617796 | de novo |
| P169 | CDH23 | 10q22.1 | c.4021G>A c.5735G>A | p.(Asp1341Asn) p.(Arg1912Gln) | Het | AR | #601067 | Maternal Paternal |
| ID | Deletion/Duplication | Location | Estimated Size | Reported Critical Genes | CNV |
|---|---|---|---|---|---|
| P6 | Deletion | 1q44 | 1.5 Kb | ZBTB18 | #612337 |
| P22 | Duplication | 9p24 | 192 Kb | DOCK8 | #614113 |
| P27 | Deletion | 7q21.3 | 8.7 Mb | COL1A2 | |
| P48 | Duplication | 2p16.1p11.2 | 32.6 MB | BCL11A | |
| P148 | Deletion | 22q11.2 | 1.9 Mb | - | #611867 |
| P176 | Duplication | 16p11.2 | 529 kb | - | #614671 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez Suárez, A.; Martínez Menéndez, B.; Escolar Escamilla, E.; Martínez Sarries, F.J.; Esparza Garrido, M.I.; Gil-Fournier, B.; Ramiro León, S.; Rubio Gribble, B.; Quesada Espinosa, J.F.; Alcaraz Romero, A.J. Whole Exome Sequencing and Panel-Based Analysis in 176 Spanish Children with Neurodevelopmental Disorders: Focus on Autism Spectrum Disorder and/or Intellectual Disability/Global Developmental Delay. Genes 2024, 15, 1310. https://doi.org/10.3390/genes15101310
Sánchez Suárez A, Martínez Menéndez B, Escolar Escamilla E, Martínez Sarries FJ, Esparza Garrido MI, Gil-Fournier B, Ramiro León S, Rubio Gribble B, Quesada Espinosa JF, Alcaraz Romero AJ. Whole Exome Sequencing and Panel-Based Analysis in 176 Spanish Children with Neurodevelopmental Disorders: Focus on Autism Spectrum Disorder and/or Intellectual Disability/Global Developmental Delay. Genes. 2024; 15(10):1310. https://doi.org/10.3390/genes15101310
Chicago/Turabian StyleSánchez Suárez, Ariadna, Beatriz Martínez Menéndez, Eduardo Escolar Escamilla, Francisco J. Martínez Sarries, Miren Iranzu Esparza Garrido, Belén Gil-Fournier, Soraya Ramiro León, Bárbara Rubio Gribble, Juan F. Quesada Espinosa, and Andrés J. Alcaraz Romero. 2024. "Whole Exome Sequencing and Panel-Based Analysis in 176 Spanish Children with Neurodevelopmental Disorders: Focus on Autism Spectrum Disorder and/or Intellectual Disability/Global Developmental Delay" Genes 15, no. 10: 1310. https://doi.org/10.3390/genes15101310