
A New Case of Mitochondrial RNA Helicase SUPV3L1-Associated Neurodegenerative Disease: Ataxia, Spasticity, Optic Atrophy, and Skin Hypopigmentation (ASOASH)
 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Molecular Genetic Methods
2.2. RNA-Analysis from Fibroblasts
2.3. Fibroblasts Cultures Preparation
2.4. Complex I Content in Fibroblast Lysates
3. Results
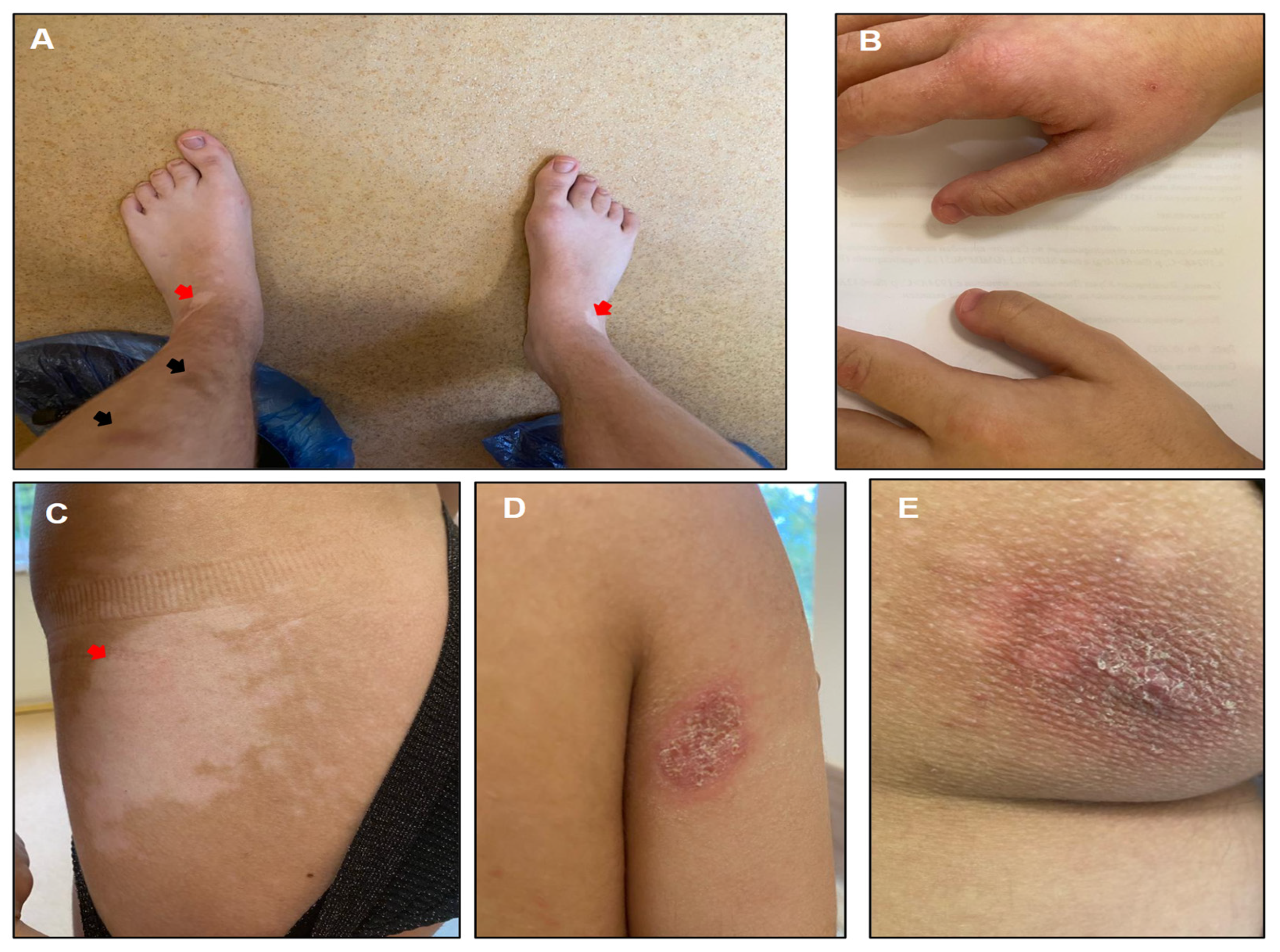
3.1. Clinical Case
3.2. Anamnesis
3.2.1. 0–7 Years
3.2.2. 7–12 Years
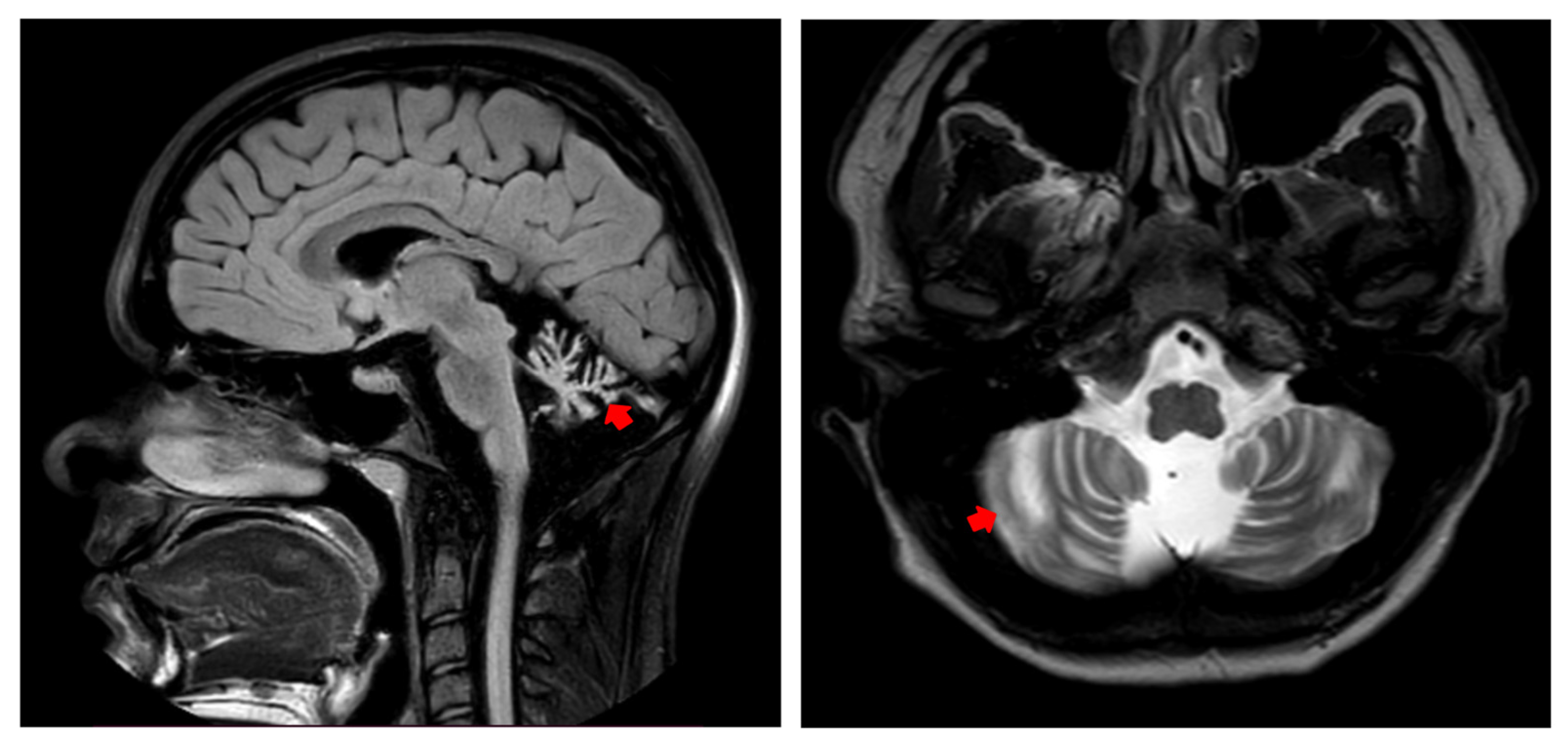
3.2.3. Evaluation at 17 Years Old
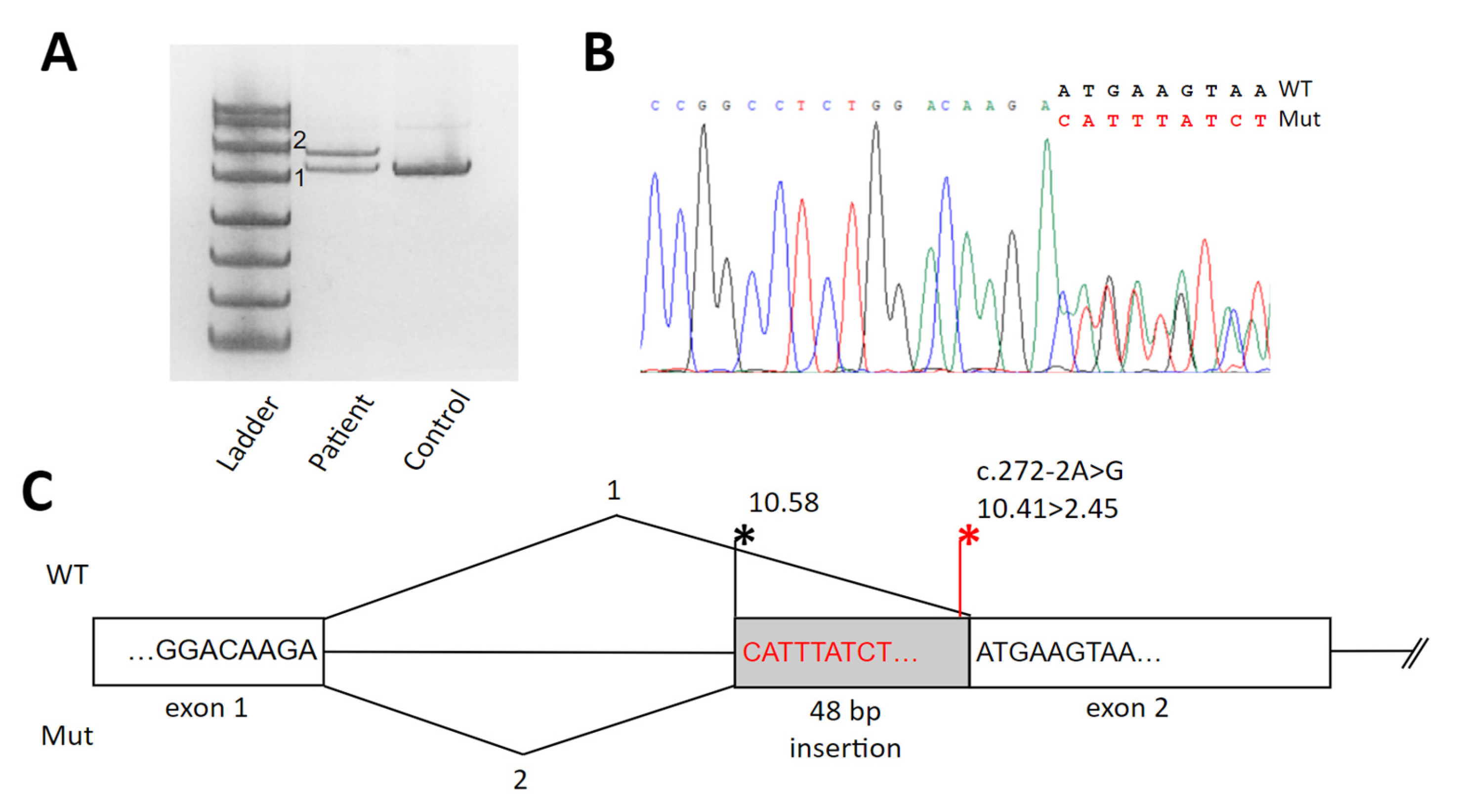
3.3. Molecular Findings
Massive Parallel Sequencing
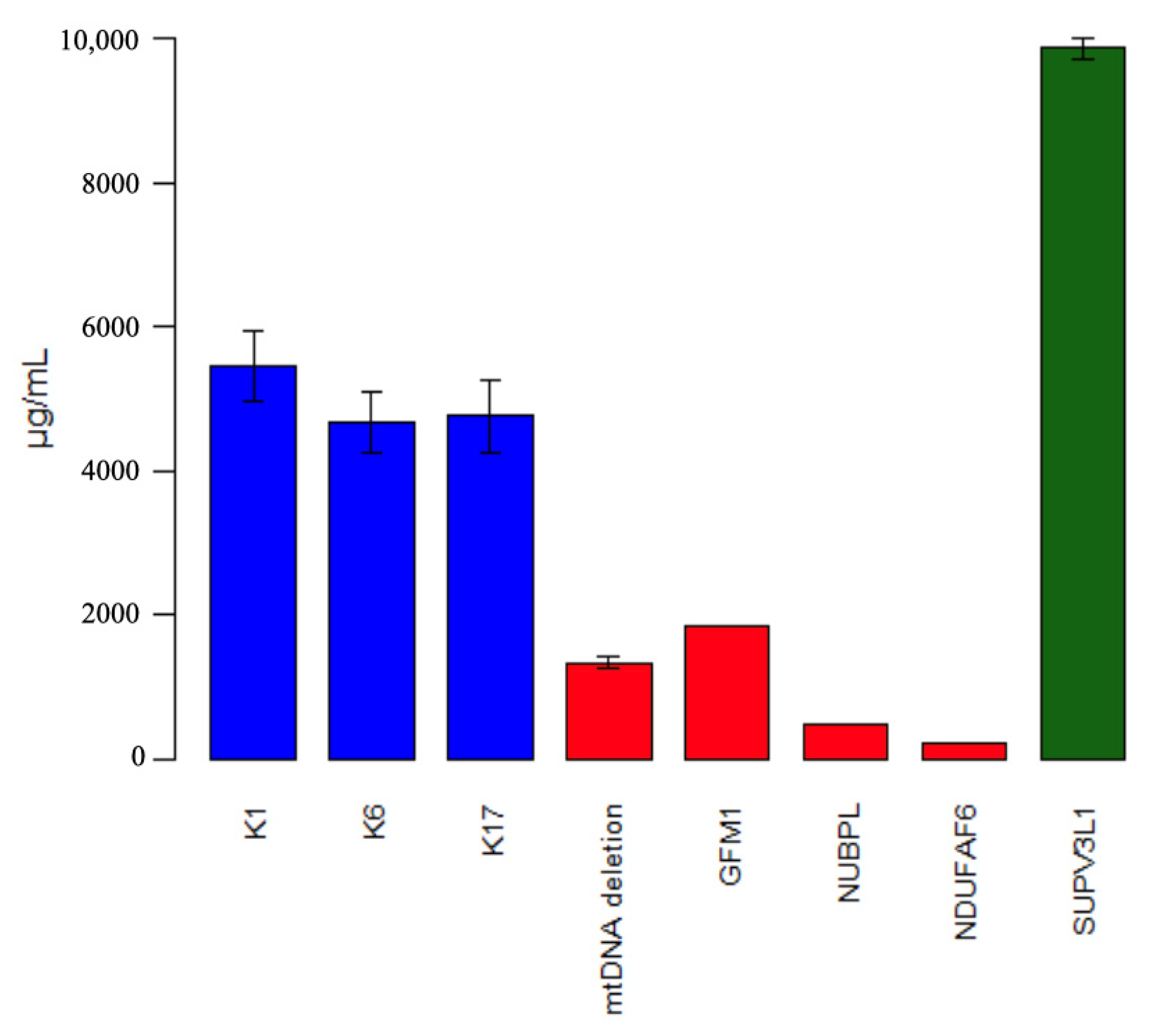
3.4. Complex 1 Content
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Borowski, L.S.; Dziembowski, A.; Hejnowicz, M.S.; Stepien, P.P.; Szczesny, R.J. Human mitochondrial RNA decay mediated by PNPase–hSuv3 complex takes place in distinct foci. Nucleic Acids Res. 2013, 41, 1223–1240. [Google Scholar] [CrossRef] [PubMed]
- Shu, Z.; Vijayakumar, S.; Chen, C.-F.; Chen, P.-L.; Lee, W.-H. Purified human SUV3p exhibits multiple-substrate unwinding activity upon conformational change. Biochemistry 2004, 43, 4781–4790. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.-H.; Shu, Z.; Lieser, S.A.; Chen, P.-L.; Lee, W.-H. Human mitochondrial SUV3 and polynucleotide phosphorylase form a 330-kDa heteropentamer to cooperatively degrade double-stranded RNA with a 3’-to-5’ directionality. J. Biol. Chem. 2009, 284, 20812–20821. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, R.J.; Borowski, L.S.; Brzezniak, L.K.; Dmochowska, A.; Gewartowski, K.; Bartnik, E.; Stepien, P.P. Human mitochondrial RNA turnover caught in flagranti: Involvement of hSuv3p helicase in RNA surveillance. Nucleic Acids Res. 2010, 38, 279–298. [Google Scholar] [CrossRef] [PubMed]
- van Esveld, S.L.; Rodenburg, R.J.; Al-Murshedi, F.; Al-Ajmi, E.; Al-Zuhaibi, S.; Huynen, M.A.; Spelbrink, J.N. Mitochondrial RNA processing defect caused by a SUPV3L1 mutation in two siblings with a novel neurodegenerative syndrome. J. Inherit. Metab. Dis. 2022, 45, 292–307. [Google Scholar] [CrossRef] [PubMed]
- MaxEntScan Online Programm for Estimation of the Strenght of the Splicing Sites in 3’ Intron Regions. Available online: http://hollywood.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq_acc.html (accessed on 1 October 2024).
- Minczuk, M.; Mroczek, S.; Pawlak, S.D.; Stepien, P.P. Human ATP-dependent RNA/DNA helicase hSuv3p interacts with the cofactor of survivin HBXIP. FEBS J. 2005, 272, 5008–5019. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Golzarroshan, B.; Lin, C.; Agrawal, S.; Tang, W.; Wu, C.; Yuan, H.S. Dimeric assembly of human Suv3 helicase promotes its RNA unwinding function in mitochondrial RNA degradosome for RNA decay. Protein Sci. 2022, 31, 4312. [Google Scholar] [CrossRef] [PubMed]
- Green, L.; Hamilton, N.; Elpidorou, M.; Maroofian, R.; Douglas, A.G.L.; Õunap, K.; Rose, A.M.S.; Harris, E.L.; Elworthy, S.; Renshaw, S.A.; et al. Biallelic Mutations in SUPV3L1 Cause an Inherited Neurodevelopmental Disorder with Variable Leukodystrophy Due to Ab-errant Mitochondrial Double Stranded RNA Processing. Unpublished. Available online: https://www.researchgate.net/publication/380513938_Biallelic_mutations_in_SUPV3L1_cause_an_inherited_neurodevelopmental_disorder_with_variable_leukodystrophy_due_to_aberrant_mitochondrial_double_stranded_RNA_processing (accessed on 10 October 2024).
- Paul, E.; Cronan, R.; Weston, P.J.; Boekelheide, K.; Sedivy, J.M.; Lee, S.-Y.; Wiest, D.L.; Resnick, M.B.; Klysik, J.E. Disruption of Supv3L1 damages the skin and causes sarcopenia, loss of fat, and death. Mamm. Genome 2009, 20, 92–108. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsygankova, P.; Chistol, D.; Krylova, T.; Bychkov, I.; Tabakov, V.; Markova, T.; Dadali, E.; Zakharova, E. A New Case of Mitochondrial RNA Helicase SUPV3L1-Associated Neurodegenerative Disease: Ataxia, Spasticity, Optic Atrophy, and Skin Hypopigmentation (ASOASH). Genes 2024, 15, 1406. https://doi.org/10.3390/genes15111406
Tsygankova P, Chistol D, Krylova T, Bychkov I, Tabakov V, Markova T, Dadali E, Zakharova E. A New Case of Mitochondrial RNA Helicase SUPV3L1-Associated Neurodegenerative Disease: Ataxia, Spasticity, Optic Atrophy, and Skin Hypopigmentation (ASOASH). Genes. 2024; 15(11):1406. https://doi.org/10.3390/genes15111406
Chicago/Turabian StyleTsygankova, Polina, Denis Chistol, Tatiana Krylova, Igor Bychkov, Vyacheslav Tabakov, Tatiana Markova, Elena Dadali, and Ekaterina Zakharova. 2024. "A New Case of Mitochondrial RNA Helicase SUPV3L1-Associated Neurodegenerative Disease: Ataxia, Spasticity, Optic Atrophy, and Skin Hypopigmentation (ASOASH)" Genes 15, no. 11: 1406. https://doi.org/10.3390/genes15111406
APA StyleTsygankova, P., Chistol, D., Krylova, T., Bychkov, I., Tabakov, V., Markova, T., Dadali, E., & Zakharova, E. (2024). A New Case of Mitochondrial RNA Helicase SUPV3L1-Associated Neurodegenerative Disease: Ataxia, Spasticity, Optic Atrophy, and Skin Hypopigmentation (ASOASH). Genes, 15(11), 1406. https://doi.org/10.3390/genes15111406