

Genome-Wide Association Studies and Runs of Homozygosity Reveals Genetic Markers Associated with Reproductive Performance in Korean Duroc, Landrace, and Yorkshire Breeds


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Phenotype
2.2. Genotyping and Quality Control
2.3. Phenotypic Correlation Analysis
2.4. Principal Component Analysis (PCA)
2.5. Genome-Wide Association Study
2.6. Identification and Annotation of Candidate Genes from GWAS
2.7. Gene Exploration of Shared ROH Regions
3. Results
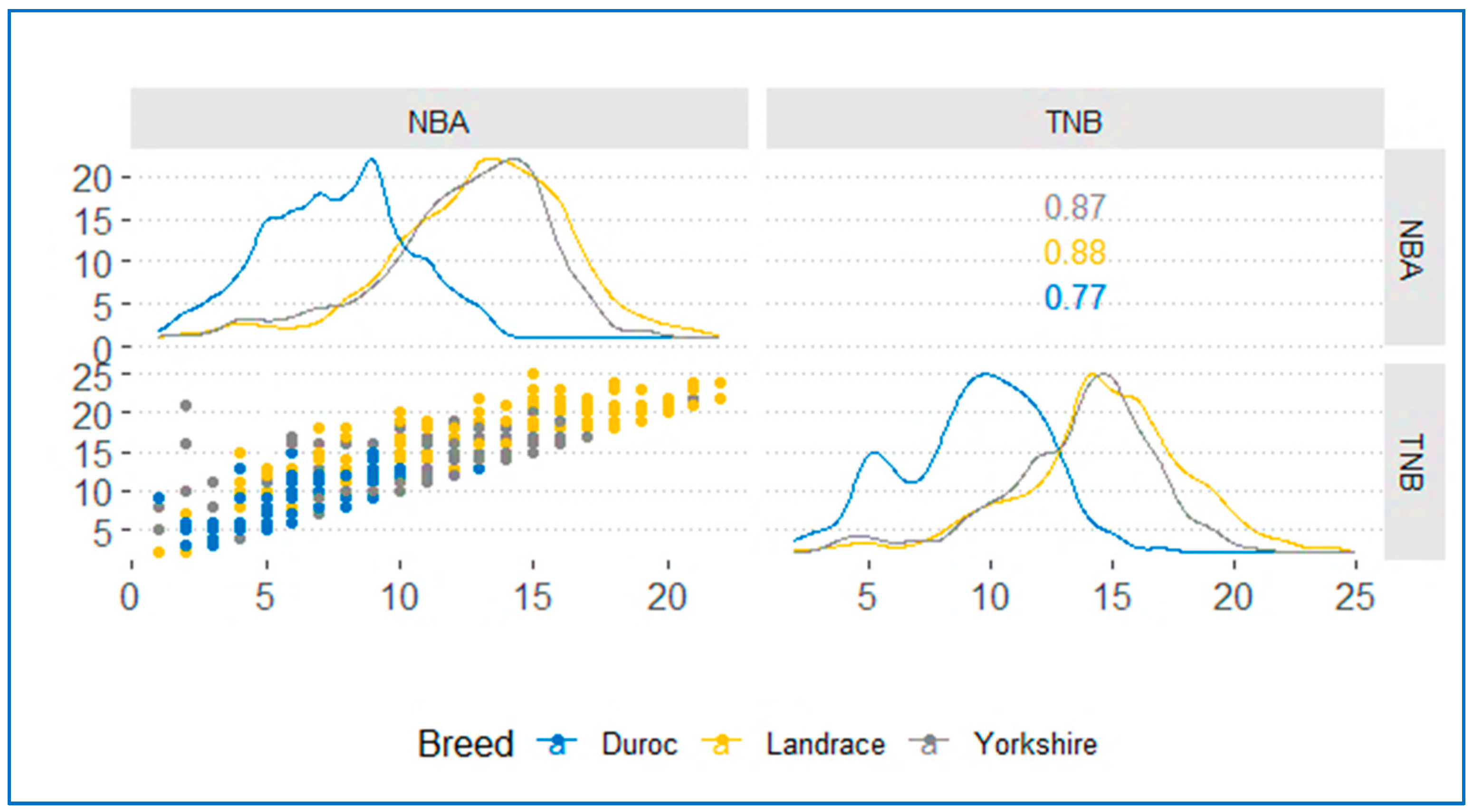
3.1. Phenotype Correlation of Reproductive Traits
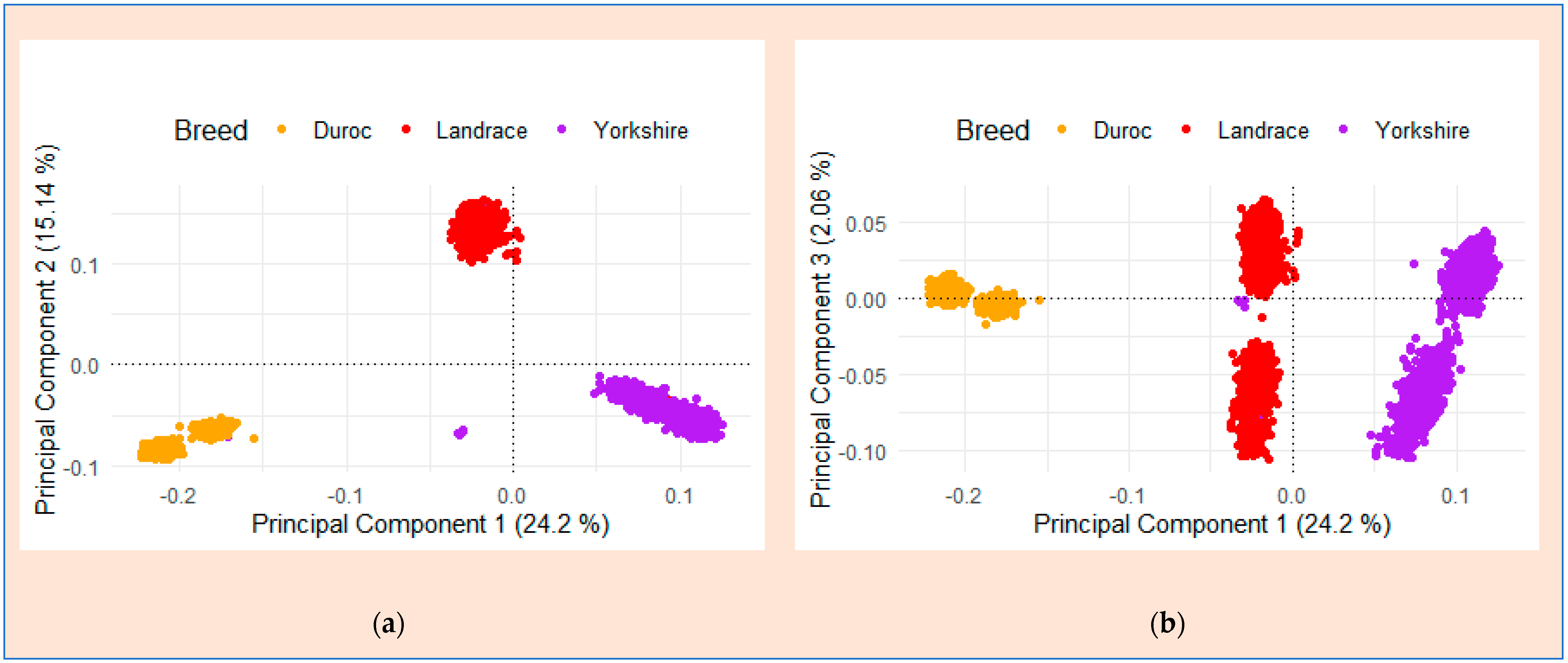
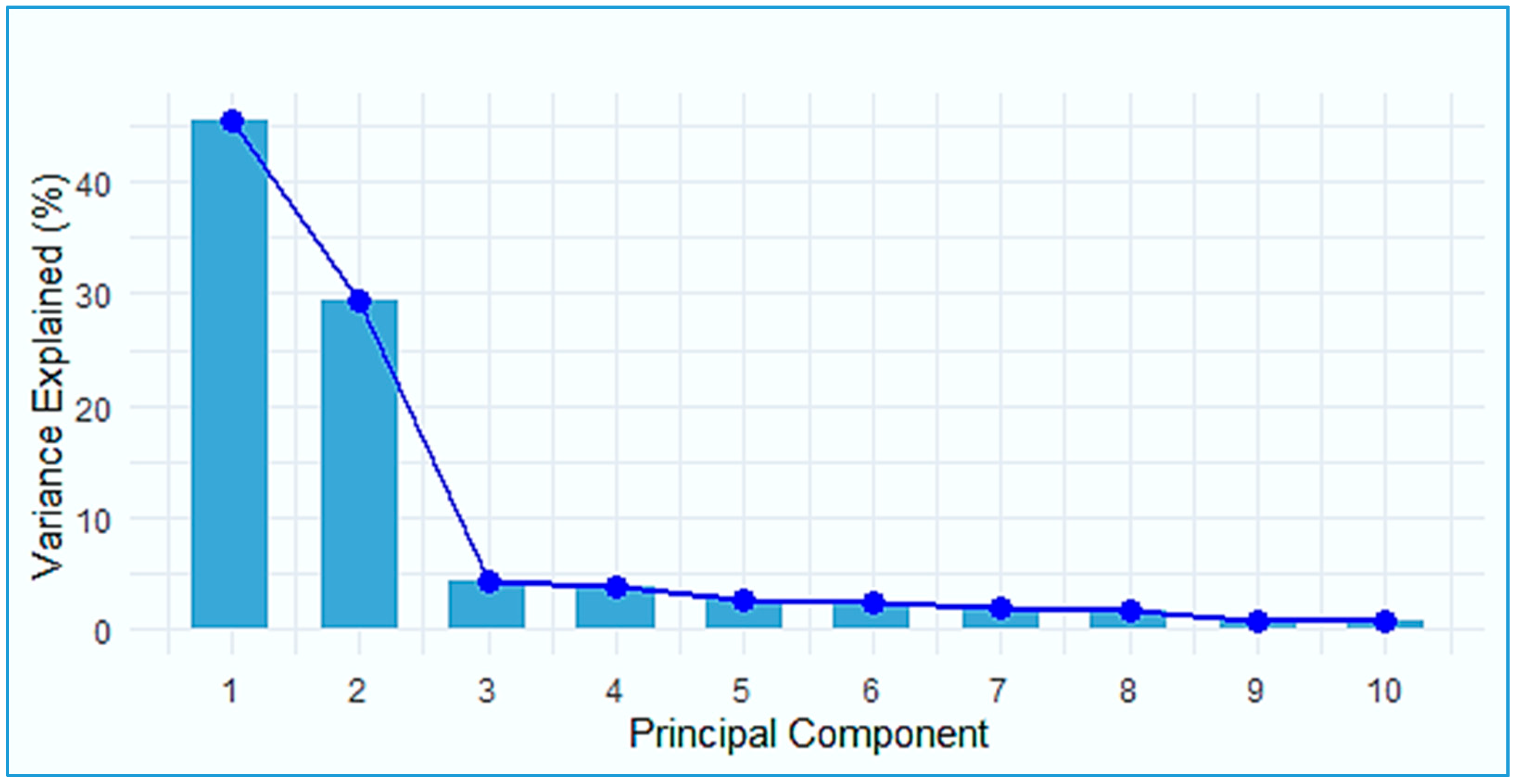
3.2. Principal Component Analysis (PCA) and Population Structure
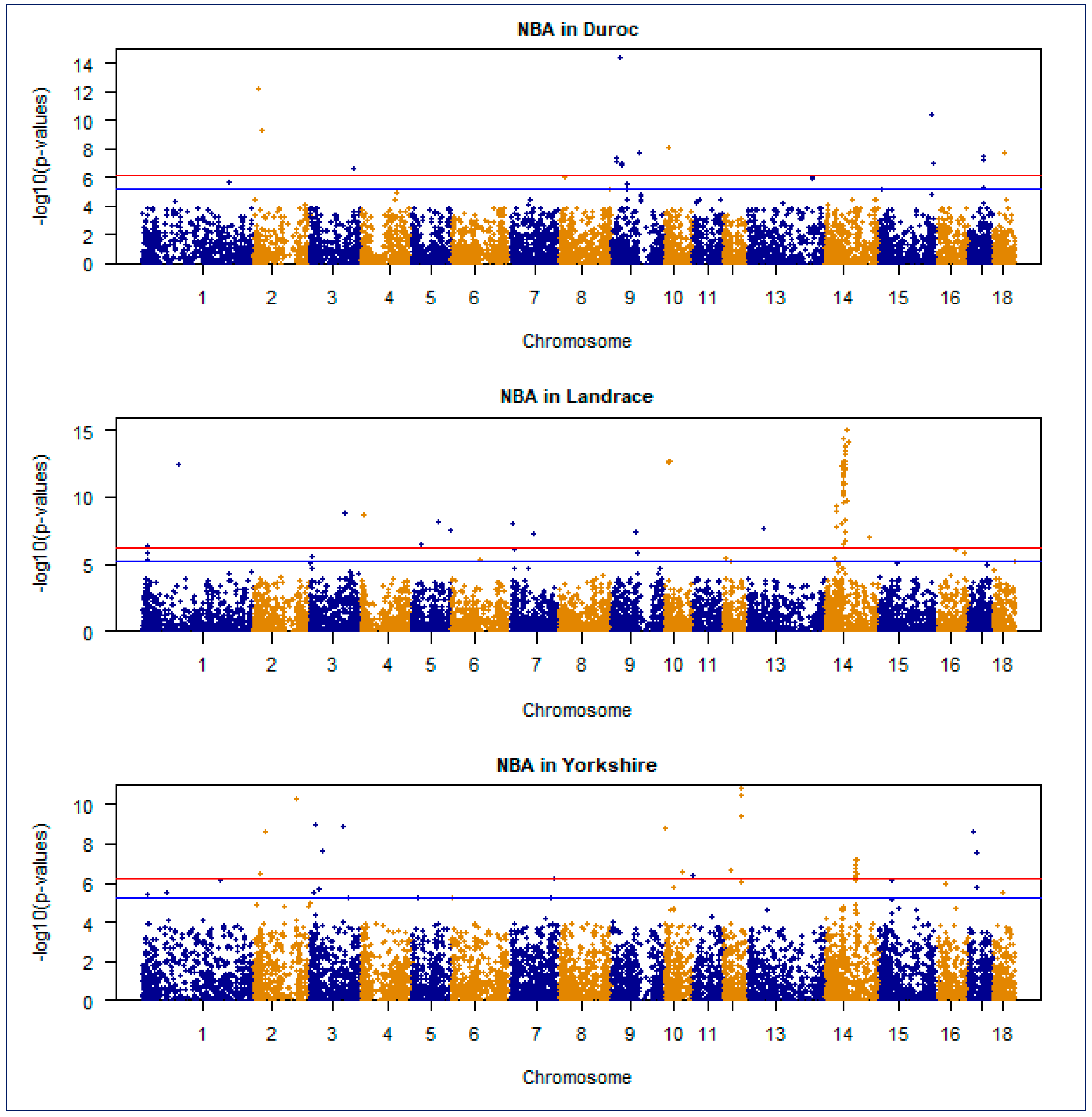
3.3. The Significant SNPs and Associated Genes from GWAS
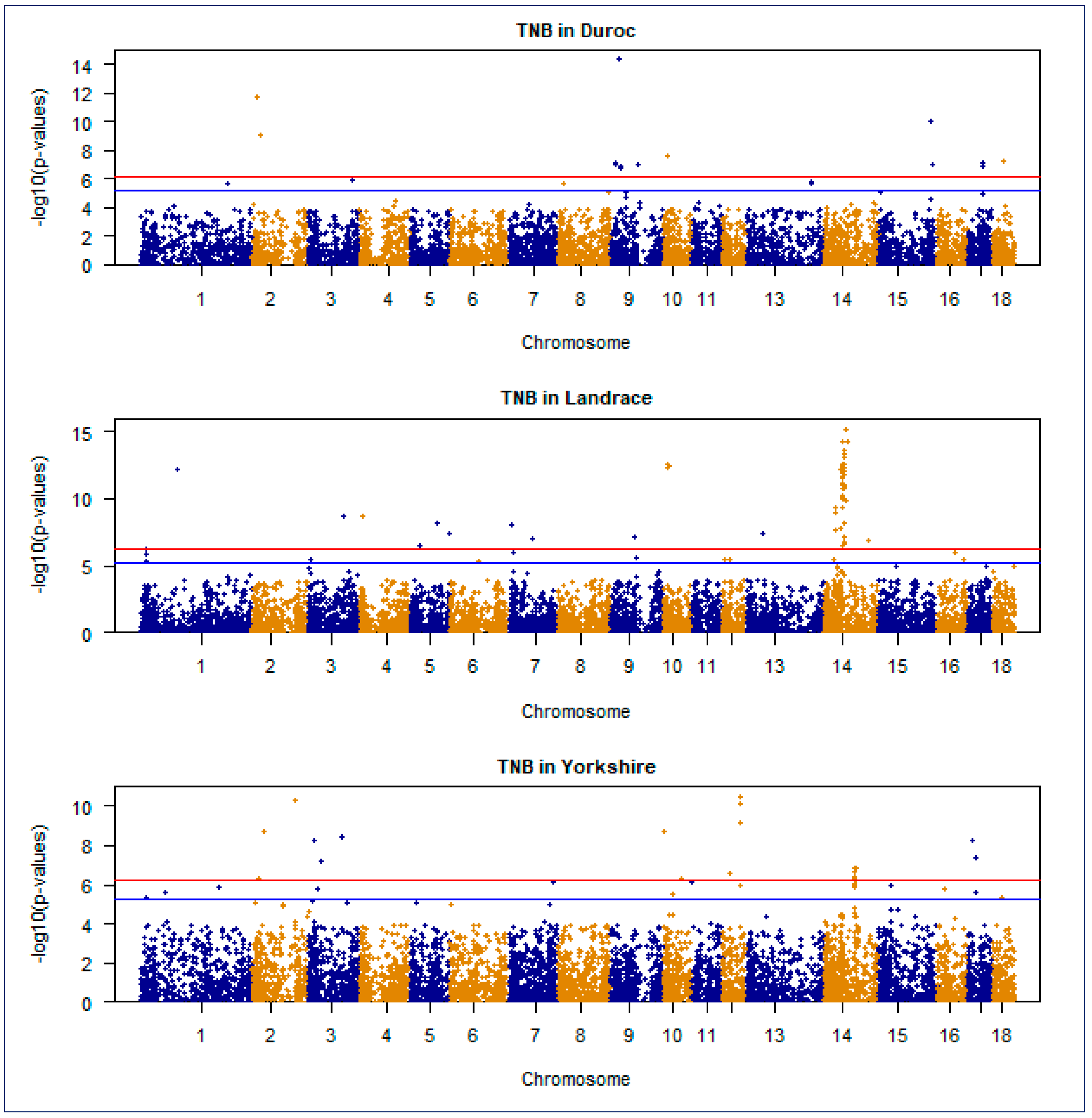
3.4. Manhattan and Quantile–Quantile Plot
3.5. GO Annotation of Candidate Genes
3.6. Genes and Their Function Within Run of Homozygosity Islands
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Onteru, S.; Fan, B.; Du, Z.Q.; Garrick, D.; Stalder, K.; Rothschild, M. A whole-genome association study for pig reproductive traits. Anim. Genet. 2012, 43, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Melo, T.P.; Fortes, M.R.; Bresolin, T.; Mota, L.F.; Albuquerque, L.G.; Carvalheiro, R. Multitrait meta-analysis identified genomic regions associated with sexual precocity in tropical beef cattle. J. Anim. Sci. 2018, 96, 4087–4099. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, M.; Jacobson, C.; Vaske, D.; Tuggle, C.; Wang, L.; Short, T.; Eckardt, G.; Sasaki, S.; Vincent, A.; McLaren, D. The estrogen receptor locus is associated with a major gene influencing litter size in pigs. Proc. Natl. Acad. Sci. USA 1996, 93, 201–205. [Google Scholar] [CrossRef]
- Fangmann, A.; Sharifi, R.; Heinkel, J.; Danowski, K.; Schrade, H.; Erbe, M.; Simianer, H. Empirical comparison between different methods for genomic prediction of number of piglets born alive in moderate sized breeding populations. J. Anim. Sci. 2017, 95, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Wang, K.; Yang, Q.; Zhou, J.; Chen, D.; Ma, J.; Tang, Q.; Jin, L.; Xiao, W.; Jiang, A. Identifying SNPs and candidate genes for three litter traits using single-step GWAS across six parities in Landrace and Large White pigs. Physiol. Genom. 2018, 50, 1026–1035. [Google Scholar] [CrossRef]
- Stranger, B.E.; Stahl, E.A.; Raj, T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics 2011, 187, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Waters, K.M.; Stram, D.O.; Hassanein, M.T.; Le Marchand, L.; Wilkens, L.R.; Maskarinec, G.; Monroe, K.R.; Kolonel, L.N.; Altshuler, D.; Henderson, B.E. Consistent association of type 2 diabetes risk variants found in europeans in diverse racial and ethnic groups. PLoS Genet. 2010, 6, e1001078. [Google Scholar] [CrossRef]
- Waters, K.M.; Le Marchand, L.; Kolonel, L.N.; Monroe, K.R.; Stram, D.O.; Henderson, B.E.; Haiman, C.A. Generalizability of associations from prostate cancer genome-wide association studies in multiple populations. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1285–1289. [Google Scholar] [CrossRef]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef]
- Wu, P.; Wang, K.; Zhou, J.; Yang, Q.; Yang, X.; Jiang, A.; Jiang, Y.; Li, M.; Zhu, L.; Bai, L. A genome wide association study for the number of animals born dead in domestic pigs. BMC Genet. 2019, 20, 4. [Google Scholar] [CrossRef]
- Sun, J.; Xiao, J.; Jiang, Y.; Wang, Y.; Cao, M.; Wei, J.; Yu, T.; Ding, X.; Yang, G. Genome-wide association study on reproductive traits using imputation-based whole-genome sequence data in Yorkshire pigs. Genes 2023, 14, 861. [Google Scholar] [CrossRef] [PubMed]
- Sell-Kubiak, E.; Knol, E.F.; Lopes, M. Evaluation of the phenotypic and genomic background of variability based on litter size of Large White pigs. Genet. Sel. Evol. 2022, 54, 1. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Tang, S.; Xiao, W.; Yun, P.; Ding, X. A genome-wide association study of reproduction traits in four pig populations with different genetic backgrounds. Asian-Australas. J. Anim. Sci. 2020, 33, 1400. [Google Scholar] [CrossRef]
- Akanno, E.; Schenkel, F.; Sargolzaei, M.; Friendship, R.; Robinson, J. Opportunities for genome-wide selection for pig breeding in developing countries. J. Anim. Sci. 2013, 91, 4617–4627. [Google Scholar] [CrossRef]
- He, S.; Di, J.; Han, B.; Chen, L.; Liu, M.; Li, W. Genome-wide scan for runs of homozygosity identifies candidate genes related to economically important traits in Chinese Merino. Animals 2020, 10, 524. [Google Scholar] [CrossRef]
- Gibson, J.; Morton, N.E.; Collins, A. Extended tracts of homozygosity in outbred human populations. Hum. Mol. Genet. 2006, 15, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Szpiech, Z.A.; Xu, J.; Pemberton, T.J.; Peng, W.; Zöllner, S.; Rosenberg, N.A.; Li, J.Z. Long runs of homozygosity are enriched for deleterious variation. Am. J. Hum. Genet. 2013, 93, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Ringbauer, H.; Novembre, J.; Steinrücken, M. Detecting runs of homozygosity from low-coverage ancient DNA. bioRxiv 2020. [Google Scholar]
- Ringbauer, H.; Novembre, J.; Steinrücken, M. Parental relatedness through time revealed by runs of homozygosity in ancient DNA. Nat. Commun. 2021, 12, 5425. [Google Scholar] [CrossRef]
- Ojeda-Marín, C.; Gutiérrez, J.P.; Formoso-Rafferty, N.; Goyache, F.; Cervantes, I. Differential patterns in runs of homozygosity in two mice lines under divergent selection for environmental variability for birth weight. J. Anim. Breed. Genet. 2024, 141, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Ciani, E.; Sardina, M.; Sottile, G.; Pilla, F.; Portolano, B.; Consortium, B.O.I. Runs of homozygosity reveal genome-wide autozygosity in Italian sheep breeds. Anim. Genet. 2018, 49, 71–81. [Google Scholar] [CrossRef]
- Xie, R.; Shi, L.; Liu, J.; Deng, T.; Wang, L.; Liu, Y.; Zhao, F. Genome-wide scan for runs of homozygosity identifies candidate genes in three pig breeds. Animals 2019, 9, 518. [Google Scholar] [CrossRef]
- Zhao, G.; Zhang, T.; Liu, Y.; Wang, Z.; Xu, L.; Zhu, B.; Gao, X.; Zhang, L.; Gao, H.; Liu, G.E. Genome-wide assessment of runs of homozygosity in Chinese wagyu beef cattle. Animals 2020, 10, 1425. [Google Scholar] [CrossRef]
- Curik, I.; Ferenčaković, M.; Sölkner, J. Inbreeding and runs of homozygosity: A possible solution to an old problem. Livest. Sci. 2014, 166, 26–34. [Google Scholar] [CrossRef]
- Dixit, S.; Singh, S.; Ganguly, I.; Bhatia, A.K.; Sharma, A.; Kumar, N.A.; Dang, A.K.; Jayakumar, S. Genome-wide runs of homozygosity revealed selection signatures in Bos indicus. Front. Genet. 2020, 11, 92. [Google Scholar] [CrossRef]
- Onzima, R.B.; Upadhyay, M.R.; Doekes, H.P.; Brito, L.F.; Bosse, M.; Kanis, E.; Groenen, M.A.; Crooijmans, R.P. Genome-wide characterization of selection signatures and runs of homozygosity in Ugandan goat breeds. Front. Genet. 2018, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Peripolli, E.; Munari, D.; Silva, M.; Lima, A.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef]
- Colpitts, J.; McLoughlin, P.D.; Poissant, J. Runs of homozygosity in Sable Island feral horses reveal the genomic consequences of inbreeding and divergence from domestic breeds. BMC Genom. 2022, 23, 501. [Google Scholar] [CrossRef]
- Islam, R.; Li, Y.; Liu, X.; Berihulay, H.; Abied, A.; Gebreselassie, G.; Ma, Q.; Ma, Y. Genome-wide runs of homozygosity, effective population size, and detection of positive selection signatures in six Chinese goat breeds. Genes 2019, 10, 938. [Google Scholar] [CrossRef]
- Brüniche-Olsen, A.; Kellner, K.F.; Anderson, C.J.; DeWoody, J.A. Runs of homozygosity have utility in mammalian conservation and evolutionary studies. Conserv. Genet. 2018, 19, 1295–1307. [Google Scholar] [CrossRef]
- Zavarez, L.B.; Utsunomiya, Y.T.; Carmo, A.S.; Neves, H.H.; Carvalheiro, R.; Ferenčaković, M.; Pérez O’Brien, A.M.; Curik, I.; Cole, J.B.; Van Tassell, C.P. Assessment of autozygosity in Nellore cows (Bos indicus) through high-density SNP genotypes. Front. Genet. 2015, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Pearson, T.A.; Manolio, T.A. How to interpret a genome-wide association study. Jama 2008, 299, 1335–1344. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, J.; Chen, C.J.; Zhang, J.; Wen, W.; Tian, J.; Zhang, Z.; Gu, Y. GWAS-based identification of new loci for milk yield, fat, and protein in Holstein cattle. Animals 2020, 10, 2048. [Google Scholar] [CrossRef]
- Andersson, L. Genome-wide association analysis in domestic animals: A powerful approach for genetic dissection of trait loci. Genetica 2009, 136, 341–349. [Google Scholar] [CrossRef]
- Wang, K.; Liu, D.; Hernandez-Sanchez, J.; Chen, J.; Liu, C.; Wu, Z.; Fang, M.; Li, N. Genome wide association analysis reveals new production trait genes in a male Duroc population. PLoS ONE 2015, 10, e0139207. [Google Scholar] [CrossRef]
- Fan, B.; Onteru, S.K.; Du, Z.-Q.; Garrick, D.J.; Stalder, K.J.; Rothschild, M.F. Genome-wide association study identifies loci for body composition and structural soundness traits in pigs. PLoS ONE 2011, 6, e14726. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luo, Y.; Fu, W.; Lu, X.; Zhou, J.; Ding, X.; Liu, J.; Zhang, Q. Genome-wide association studies for hematological traits in swine. Anim. Genet. 2013, 44, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Bouaziz, M.; Ambroise, C.; Guedj, M. Accounting for population stratification in practice: A comparison of the main strategies dedicated to genome-wide association studies. PLoS ONE 2011, 6, e28845. [Google Scholar] [CrossRef]
- Geng, M.-y.; Wang, L.; Song, Y.-y.; Gu, J.; Hu, X.; Yuan, C.; Yang, M.; Pei, W.-j.; Zhang, Y.; Gao, J.-l. Sidt2 is a key protein in the autophagy-lysosomal degradation pathway and is essential for the maintenance of kidney structure and filtration function. Cell Death Dis. 2021, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wu, M.; Wu, X.; Chen, D.; Xie, M.; Pan, H. TGM2 accelerates migration and differentiation of BMSCs by activating Wnt/β-catenin signaling. J. Orthop. Surg. Res. 2023, 18, 168. [Google Scholar] [CrossRef]
- Chang, W.; Gao, W.; Liu, D.; Luo, B.; Li, H.; Zhong, L.; Chen, Y. The upregulation of TGM2 is associated with poor prognosis and the shaping of the inflammatory tumor microenvironment in lung squamous cell carcinoma. Am. J. Cancer Res. 2024, 14, 2823. [Google Scholar] [CrossRef] [PubMed]
- Mchta, K. Mammalian transglutaminases: A family portrait. Prog. Exp. Tumor Res. 2005, 38, 5–7. [Google Scholar]
- Agnihotri, N.; Kumar, S.; Mehta, K. Tissue transglutaminase as a central mediator in inflammation-induced progression of breast cancer. Breast Cancer Res. 2013, 15, 202. [Google Scholar] [CrossRef]
- Bencze, J.; Mórotz, G.M.; Seo, W.; Bencs, V.; Kálmán, J.; Miller, C.C.J.; Hortobágyi, T. Biological function of Lemur tyrosine kinase 2 (LMTK2): Implications in neurodegeneration. Mol. Brain 2018, 11, 20. [Google Scholar] [CrossRef]
- Clavijo, R.I.; Arora, H.; Gibbs, E.; Cohen, S.; Griswold, A.; Bakircioglu, E.; Bademci, G.; Tekin, M.; Ramasamy, R. Whole exome sequencing of a consanguineous Turkish family identifies a mutation in GTF2H3 in brothers with spermatogenic failure. Urology 2018, 120, 86–89. [Google Scholar] [CrossRef]
- Ye, J.; Li, X.; Pan, Z.; Wu, Z.; Zhu, Y.; Zhang, W.; Lu, J.; Xu, S.; Qin, P.; Liu, Y. Grid1 regulates the onset of puberty in female rats. J. Vet. Med. Sci. 2024, 86, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Tencer, A.; Xuan, H.; Kutateladze, T.G.; Shi, X. ZZEF1 is a histone reader and transcriptional coregulator of Krüppel-like factors. J. Mol. Biol. 2021, 433, 166722. [Google Scholar] [CrossRef]
- Wasik, K.A.; Tam, O.H.; Knott, S.R.; Falciatori, I.; Hammell, M.; Vagin, V.V.; Hannon, G.J. RNF17 blocks promiscuous activity of PIWI proteins in mouse testes. Genes Dev. 2015, 29, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Mekonnen, K.T.; Lee, D.-H.; Cho, Y.-G.; Son, A.-Y.; Seo, K.-S. Genomic and Conventional Inbreeding Coefficient Estimation Using Different Estimator Models in Korean Duroc, Landrace, and Yorkshire Breeds Using 70K Porcine SNP BeadChip. Animals 2024, 14, 2621. [Google Scholar] [CrossRef] [PubMed]
- Rachev, E.; Schuster-Gossler, K.; Fuhl, F.; Ott, T.; Tveriakhina, L.; Beckers, A.; Hegermann, J.; Boldt, K.; Mai, M.; Kremmer, E. CFAP43 modulates ciliary beating in mouse and Xenopus. Dev. Biol. 2020, 459, 109–125. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, J.; Zhou, L.; Li, H.; Zheng, B.; Yang, S. CFAP43-mediated intra-manchette transport is required for sperm head shaping and flagella formation. Zygote 2021, 29, 75–81. [Google Scholar] [CrossRef]
- Jin, Z.; Zhao, L.; Chang, Y.; Jin, R.; Hu, F.; Wu, S.; Xue, Z.; Ma, Y.; Chen, C.; Zheng, M. CRTAC1 enhances the chemosensitivity of non-small cell lung cancer to cisplatin by eliciting RyR-mediated calcium release and inhibiting Akt1 expression. Cell Death Dis. 2023, 14, 563. [Google Scholar] [CrossRef]
- Rademacher, D.J.; Bello, A.I.; May, J.P. CASC3 Biomolecular Condensates Restrict Turnip Crinkle Virus by Limiting Host Factor Availability. J. Mol. Biol. 2023, 435, 167956. [Google Scholar] [CrossRef] [PubMed]
- Gerbracht, J.V.; Boehm, V.; Britto-Borges, T.; Kallabis, S.; Wiederstein, J.L.; Ciriello, S.; Aschemeier, D.U.; Krüger, M.; Frese, C.K.; Altmüller, J. CASC3 promotes transcriptome-wide activation of nonsense-mediated decay by the exon junction complex. Nucleic Acids Res. 2020, 48, 8626–8644. [Google Scholar] [CrossRef]
- Woodley, K.T.; Collins, M.O. S-acylated Golga7b stabilises DHHC 5 at the plasma membrane to regulate cell adhesion. EMBO Rep. 2019, 20, e47472. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | Duroc | Landrace | Yorkshire | |||
|---|---|---|---|---|---|---|
| n | Mean ± SD | n | Mean ± SD | n | Mean ± SD | |
| NBA | 1162 | 7.60 ± 2.65 | 1782 | 12.42 ± 3.11 | 2363 | 13.06 ± 3.27 |
| TNB | 1162 | 9.26 ± 2.92 | 1783 | 13.61 ± 3.20 | 2364 | 14.61 ± 3.52 |
| Trait | SSC | SNP | Position | p-Value | Allele | β | Distance | Gene | Gene ID |
|---|---|---|---|---|---|---|---|---|---|
| TNB | 13 | Affx-115053386 | 181,341,264 | 1.52 × 10−6 | G/A | 12.11 | within | Unknown | ENSSSCG00000063239 |
| TNB | 8 | WU_10.2_8_18342200 | 18,342,200 | 2.01 × 10−6 | A/C | −10.84 | within | Unknown | ENSSSCG00000056996 |
| NBA | 8 | WU_10.2_8_18342200 | 18,342,200 | 8.68 × 10−7 | A/C | −6.498 | within | Unknown | ENSSSCG00000056996 |
| NBA | 13 | Affx-115053386 | 181,341,264 | 9.56 × 10−7 | G/A | 6.483 | 131 | Unknown | ENSSSCG00000063239 |
| NBA | 9 | DRGA0009521 | 44,552,360 | 3.23 × 10−6 | G/A | −6.292 | within | SIDT2 | ENSSSCG00000015074 |
| NBA | 17 | ASGA0076732 | 41,226,448 | 5.07 × 10−6 | G/A | 6.22 | 4811 | TGM2 | ENSSSCG00000023522 |
| Traits | SSC | SNP | Position | p-Value | Allele | β | Distance | Genes | Gene ID |
|---|---|---|---|---|---|---|---|---|---|
| TNB | 1 | WU_10.2_1_13788588 | 13,788,588 | 1.35 × 10−6 | G/A | 8.778 | within | Unknown | ENSSSCG00000004081 |
| TNB | 9 | ALGA0053627 | 74,677,600 | 2.33 × 10−6 | A/G | −8.525 | within | PPP1R9A | ENSSSCG00000015329 |
| TNB | 3 | ALGA0017853 | 5,346,840 | 3.32 × 10−6 | A/G | −12.24 | within | LMTK2 | ENSSSCG00000007599 |
| TNB | 14 | ALGA0076573 | 29,354,768 | 3.38 × 10−6 | A/C | −8.139 | within | GTF2H3 | ENSSSCG00000009769 |
| NBA | 1 | WU_10.2_1_13788588 | 13,788,588 | 1.34 × 10−6 | G/A | 8.65 | within | Unknown | ENSSSCG00000004081 |
| NBA | 9 | ALGA0053627 | 74,677,600 | 1.55 × 10−6 | A/G | −8.483 | within | PPP1R9A | ENSSSCG00000015329 |
| NBA | 3 | ALGA0017853 | 5,346,840 | 2.37 × 10−6 | A/G | −12.17 | within | LMTK2 | ENSSSCG00000007599 |
| NBA | 14 | ALGA0076573 | 29,354,768 | 3.06 × 10−6 | A/C | −8.039 | within | GTF2H3 | ENSSSCG00000009769 |
| Trait | SSC | SNP | Position | p-Value | Allele | β | Distance | Genes | Gene ID |
|---|---|---|---|---|---|---|---|---|---|
| NBA | 14 | DRGA0014176 | 86,922,816 | 6.43 × 10−7 | A/G | 9.944 | within | GRID1 | ENSSSCG00000010352 |
| NBA | 15 | WU_10.2_15_32958696 | 32,958,696 | 6.68 × 10−7 | G/A | 8.485 | within | DLGAP2 | ENSSSCG00000015746 |
| NBA | 14 | DRGA0014177 | 86,969,008 | 6.68 × 10−7 | A/C | 9.935 | within | GRID1 | ENSSSCG00000010352 |
| NBA | 1 | MARC0036130 | 218,896,192 | 7.39 × 10−7 | A/G | −15.73 | within | Unknown | ENSSSCG00000039823 |
| NBA | 12 | H3GA0034587 | 50,146,896 | 8.41 × 10−7 | G/A | −18.33 | within | ZZEF1 | ENSSSCG00000032049 |
| NBA | 10 | WU_10.2_10_23632408 | 23,632,408 | 1.58 × 10−6 | G/A | −9.205 | within | Unknown | ENSSSCG00000036948 |
| NBA | 17 | H3GA0048059 | 22,512,720 | 1.80 × 10−6 | A/G | −9.605 | 3838 | SEL1L2 | ENSSSCG00000007078 |
| NBA | 1 | ASGA0003159 | 67,590,048 | 2.86 × 10−6 | G/A | −8.503 | within | ASCC3 | ENSSSCG00000004358 |
| NBA | 18 | WU_10.2_18_26226731 | 26,226,732 | 3.21 × 10−6 | A/G | −12.03 | within | KCND2 | ENSSSCG00000016621 |
| NBA | 3 | ASGA0014253 | 11,933,535 | 3.36 × 10−6 | A/G | 9.992 | within | RCC1L | ENSSSCG00000007723 |
| NBA | 1 | WU_10.2_1_14946807 | 14,946,807 | 3.56 × 10−6 | A/G | −9.535 | within | AKAP12 | ENSSSCG00000031733 |
| NBA | 3 | ASGA0097775 | 107,125,992 | 5.56 × 10−6 | A/G | −8.23 | within | BIRC6 | ENSSSCG00000008513 |
| NBA | 5 | ASGA0024707 | 15,346,437 | 5.66 × 10−6 | A/G | 8.811 | 1196 | SPATS2 | ENSSSCG00000000199 |
| Trait | SSC | SNP | Position | p-Value | Allele | β | Distance | Genes | Gene ID |
|---|---|---|---|---|---|---|---|---|---|
| TNB | 14 | ASGA0064844 | 90,362,768 | 6.20× 10−7 | A/C | 10.05 | within | PARG | ENSSSCG00000010396 |
| TNB | 11 | MARC0024643 | 173,494 | 7.57× 10−7 | A/G | −19.93 | within | RNF17 | ENSSSCG00000009572 |
| TNB | 14 | MARC0088303 | 86,898,064 | 9.56× 10−7 | A/G | 10.01 | within | GRID1 | ENSSSCG00000010352 |
| TNB | 15 | WU_10.2_15_32958696 | 32,958,696 | 1.03 × 10−-6 | G/A | 8.489 | within | DLGAP2 | ENSSSCG00000015746 |
| TNB | 12 | H3GA0034587 | 50,146,896 | 1.11 × 10−6 | G/A | −18.4 | within | ZZEF1 | ENSSSCG00000032049 |
| TNB | 14 | DRGA0014176 | 86,922,816 | 1.22 × 10−6 | A/G | 9.901 | within | GRID1 | ENSSSCG00000010352 |
| TNB | 14 | DRGA0014177 | 86,969,008 | 1.27 × 10−6 | A/C | 9.892 | within | GRID1 | ENSSSCG00000010352 |
| TNB | 1 | MARC0036130 | 218,896,192 | 1.33 × 10−6 | A/G | −15.68 | within | Unknown | ENSSSCG00000039823 |
| TNB | 17 | H3GA0048059 | 22,512,720 | 2.47 × 10−6 | A/G | −9.632 | 3939 | NDUFAF5 | ENSSSCG00000007076 |
| TNB | 1 | ASGA0003159 | 67,590,048 | 2.67 × 10−6 | G/A | −8.608 | within | ASCC3 | ENSSSCG00000004358 |
| TNB | 10 | WU_10.2_10_23632408 | 23,632,408 | 2.77 × 10−6 | G/A | −9.178 | within | Unknown | ENSSSCG00000036948 |
| TNB | 18 | WU_10.2_18_26226731 | 26,226,732 | 4.17 × 10−6 | A/G | −12.07 | within | KCND2 | ENSSSCG00000016621 |
| TNB | 1 | WU_10.2_1_14946807 | 14,946,807 | 5.08 × 10−6 | A/G | −9.55 | within | AKAP12 | ENSSSCG00000031733 |
| Breed | Gene ID | Genes | SSC | Start (bp) | End (bp) | Pos (Mbp) | Function |
|---|---|---|---|---|---|---|---|
| DD | ENSSSCG00000004435 | NT5DC1 | 1 | 81,746,022 | 81,881,113 | 81.75 | Metaphyseal Chondrodysplasia |
| DD | ENSSSCG00000010530 | CRTAC1 | 14 | 109,223,293 | 109,390,691 | 109.22 | Ion binding |
| DD | ENSSSCG00000010609 | CFAP43 | 14 | 115,073,366 | 115,174,043 | 115.07 | Spermatogenic failure 19 |
| DD | ENSSSCG00000017477 | CASC3 | 12 | 22,199,228 | 22,221,499 | 22.20 | Intracellular mRNA localization |
| DD | ENSSSCG00000024417 | ERC2 | 13 | 37,388,517 | 38,367,504 | 37.39 | Cellular component organization |
| DD | ENSSSCG00000040359 | GOLGA7B | 14 | 109,210,866 | 109,225,153 | 109.21 | Golgin A7 family member B |
| LL | ENSSSCG00000028406 | FOCAD | 1 | 201,942,463 | 202,258,794 | 201.88 | Focadhesin |
| YY | ENSSSCG00000016279 | PDE6D | 15 | 132,375,743 | 132,426,683 | 132.38 | GTPase inhibitor activity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mekonnen, K.T.; Lee, D.-H.; Cho, Y.-G.; Son, A.-Y.; Seo, K.-S. Genome-Wide Association Studies and Runs of Homozygosity Reveals Genetic Markers Associated with Reproductive Performance in Korean Duroc, Landrace, and Yorkshire Breeds. Genes 2024, 15, 1422. https://doi.org/10.3390/genes15111422
Mekonnen KT, Lee D-H, Cho Y-G, Son A-Y, Seo K-S. Genome-Wide Association Studies and Runs of Homozygosity Reveals Genetic Markers Associated with Reproductive Performance in Korean Duroc, Landrace, and Yorkshire Breeds. Genes. 2024; 15(11):1422. https://doi.org/10.3390/genes15111422
Chicago/Turabian StyleMekonnen, Kefala Taye, Dong-Hui Lee, Young-Gyu Cho, Ah-Yeong Son, and Kang-Seok Seo. 2024. "Genome-Wide Association Studies and Runs of Homozygosity Reveals Genetic Markers Associated with Reproductive Performance in Korean Duroc, Landrace, and Yorkshire Breeds" Genes 15, no. 11: 1422. https://doi.org/10.3390/genes15111422
APA StyleMekonnen, K. T., Lee, D.-H., Cho, Y.-G., Son, A.-Y., & Seo, K.-S. (2024). Genome-Wide Association Studies and Runs of Homozygosity Reveals Genetic Markers Associated with Reproductive Performance in Korean Duroc, Landrace, and Yorkshire Breeds. Genes, 15(11), 1422. https://doi.org/10.3390/genes15111422