

The Complexity of Familial Inheritance in Pectus Excavatum: A Ten-Family Exome Sequencing Analysis

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
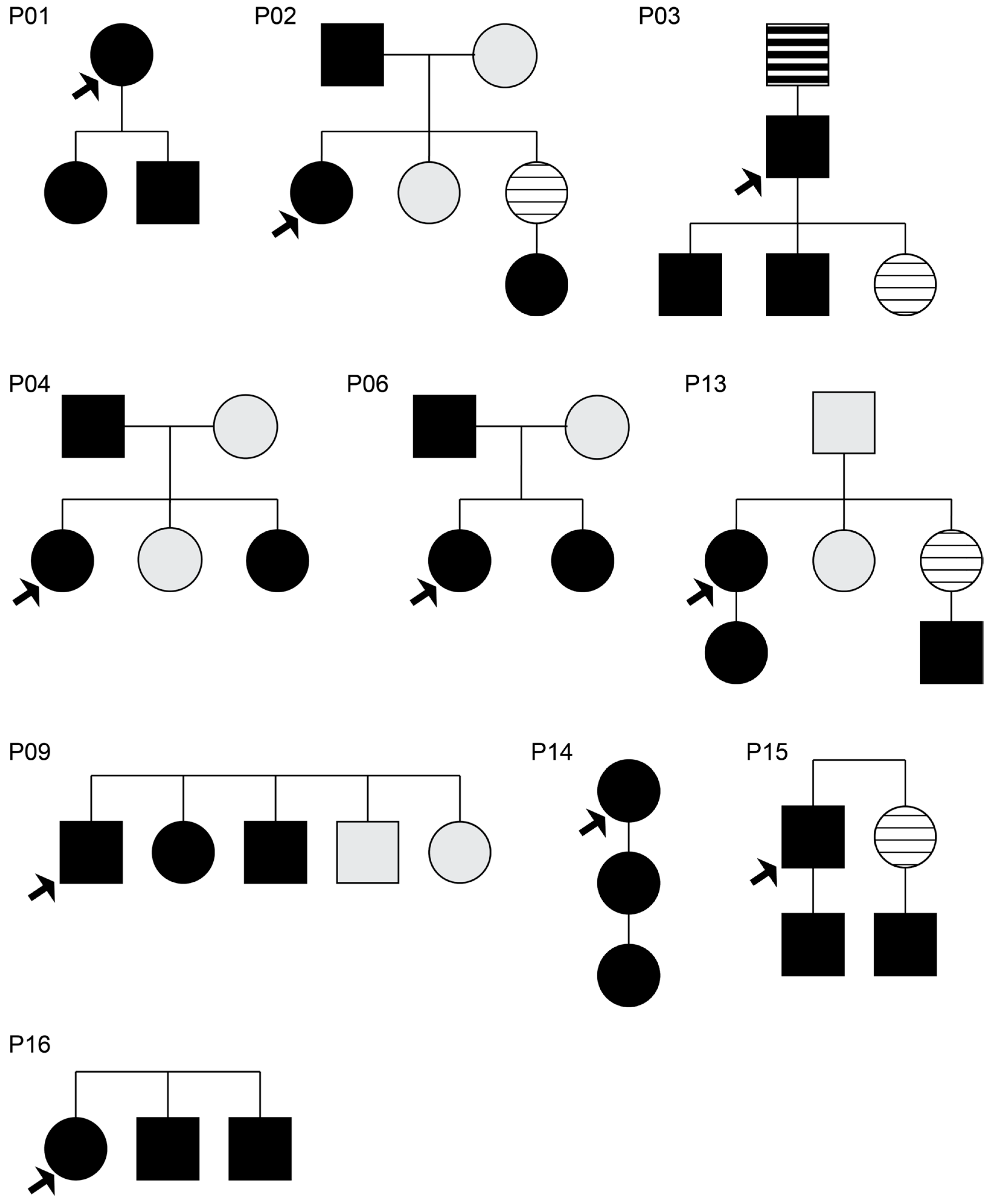
3.1. Monogenic Analysis of Affected Familial Cases
3.2. Segregation Analysis of Affected and Unaffected Family Members
3.2.1. Family P02
3.2.2. Family P04
3.2.3. Family P06, Family P09, and Family P13
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shamberger, R.C. Congenital chest wall deformities. Curr. Probl. Surg. 1996, 33, 469–542. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.S.; Myrianthopoulos, N.C. Factors affecting risks of congenital malformations. I. Analysis of epidemiologic factors in congenital malformations. Report from the Collaborative Perinatal Project. Birth Defects Orig. Artic. Ser. 1975, 11, 1–22. [Google Scholar] [PubMed]
- Creswick, H.A.; Stacey, M.W.; Kelly, R.E., Jr.; Gustin, T.; Nuss, D.; Harvey, H.; Goretsky, M.J.; Vasser, E.; Welch, J.C.; Mitchell, K.; et al. Family study of the inheritance of pectus excavatum. J. Pediatr. Surg. 2006, 41, 1699–1703. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Sun, X.; Zhu, W.; Huang, Y.; Mou, L.; Liu, M.; Li, X.; Li, F.; Li, X.; Zhang, Y.; et al. Evidence for GAL3ST4 mutation as the potential cause of pectus excavatum. Cell Res. 2012, 22, 1712–1715. [Google Scholar] [CrossRef] [PubMed]
- Horth, L.; Stacey, M.W.; Proud, V.K.; Segna, K.; Rutherford, C.; Nuss, D.; Kelly, R.E. Advancing our understanding of the inheritance and transmission of pectus excavatum. J. Pediatr. Genet. 2012, 1, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Li, G.; Feng, Y. TINAG mutation as a genetic cause of pectus excavatum. Med. Hypotheses 2020, 137, 109557. [Google Scholar] [CrossRef] [PubMed]
- Haller, J.A., Jr.; Kramer, S.S.; Lietman, S.A. Use of CT scans in selection of patients for pectus excavatum surgery: A preliminary report. J. Pediatr. Surg. 1987, 22, 904–906. [Google Scholar] [CrossRef] [PubMed]
- Funari, V.A.; Day, A.; Krakow, D.; Cohn, Z.A.; Chen, Z.; Nelson, S.F.; Cohn, D.H. Cartilage-selective genes identified in genome-scale analysis of non-cartilage and cartilage gene expression. BMC Genom. 2007, 8, 165. [Google Scholar] [CrossRef] [PubMed]
- MGI. Gene Ontology Annotations—Cartilage Development. 2023. Available online: https://www.informatics.jax.org/go/term/GO:0051216#myDataTable=results%3D100%26startIndex%3D0%26sort%3Dterm%26dir%3Dasc (accessed on 1 June 2024).
- Invitae Aortopathy Comprehensive Panel. Updated 2022-10-11. 2023. Available online: https://www.ncbi.nlm.nih.gov/gtr/tests/528496/methodology/ (accessed on 1 December 2023).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.; Pujar, S.; Loveland, J.E.; Astashyn, A.; Bennett, R.; Berry, A.; Cox, E.; Davidson, C.; Ermolaeva, O.; Farrell, C.M.; et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 2022, 604, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Karsenty, G.; Wagner, E.F. Reaching a genetic and molecular understanding of skeletal development. Dev. Cell 2002, 2, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Bayram, Y.; White, J.J.; Elcioglu, N.; Cho, M.T.; Zadeh, N.; Gedikbasi, A.; Palanduz, S.; Ozturk, S.; Cefle, K.; Kasapcopur, O.; et al. REST Final-Exon-Truncating Mutations Cause Hereditary Gingival Fibromatosis. Am. J. Hum. Genet. 2017, 101, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Manyisa, N.; Schrauwen, I.; de Souza Rios, L.A.; Mowla, S.; Tekendo-Ngongang, C.; Popel, K.; Esoh, K.; Bharadwaj, T.; Nouel-Saied, L.M.; Acharya, A.; et al. A Monoallelic Variant in REST Is Associated with Non-Syndromic Autosomal Dominant Hearing Impairment in a South African Family. Genes 2021, 12, 1765. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Kelly, M.C.; Rehman, A.U.; Boger, E.T.; Morell, R.J.; Kelley, M.W.; Friedman, T.B.; Banfi, B. Defects in the Alternative Splicing-Dependent Regulation of REST Cause Deafness. Cell 2018, 174, 536–548.e21. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.Y.; Guo, D.C.; Regalado, E.S.; Shen, H.; University of Washington Center for Mendelian, G.; Coselli, J.S.; Estrera, A.L.; Safi, H.J.; Bamshad, M.J.; Nickerson, D.A.; et al. SMAD4 rare variants in individuals and families with thoracic aortic aneurysms and dissections. Eur. J. Hum. Genet. 2019, 27, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Kloth, K.; Klohs, S.; Bhullar, J.; Boettcher, M.; Hempel, M.; Trah, J.; Reinshagen, K. The Epidemiology behind Pectus Excavatum: Clinical Study and Review of the Literature. Eur. J. Pediatr. Surg. 2022, 32, 316–320. [Google Scholar] [CrossRef] [PubMed]
- David, V.L. Current Concepts in the Etiology and Pathogenesis of Pectus Excavatum in Humans-A Systematic Review. J. Clin. Med. 2022, 11, 1241. [Google Scholar] [CrossRef] [PubMed]
- Billar, R.J.; Manoubi, W.; Kant, S.G.; Wijnen, R.M.H.; Demirdas, S.; Schnater, J.M. Association between pectus excavatum and congenital genetic disorders: A systematic review and practical guide for the treating physician. J. Pediatr. Surg. 2021, 56, 2239–2252. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Patients | Brief Clinical Presentation and Course |
|---|---|
| Proband 01 | This is a 57-year-old female who was found to have a mildly symptomatic pectus excavatum deformity with right heart compression. Patient’s CT findings revealed a Haller Index of 3.25, which worsens to 3.43 with expiration. |
| Proband 02 | This is a 40-year-old female with a history of severe and symptomatic pectus excavatum (Haller Index 3.8) who underwent minimally invasive repair. The patient underwent bar removal 3 years following initial repair. |
| Proband 03 | This is a 47-year-old male with a history of severe symptomatic pectus with a Haller Index of 3.51 and evidence of right heart compression. The patient underwent a minimally invasive repair of pectus excavatum followed by bar removal after 3 years. |
| Proband 04 | This is a 19-year-old female with a history of severe symptomatic pectus excavatum with a Haller Index of 6.8 who underwent minimally invasive repair of PEx. She underwent bar removal after 3 years with no complications. |
| Proband 06 | This is a 49-year-old female with a history of severe symptomatic pectus with a Haller Index of 5.5 and evidence of right heart compression. The patient underwent minimally invasive repair of pectus excavatum followed by bar removal and bone osteophyte excision after 3 years. |
| Proband 09 | This is a 53-year-old male with a history of severe symptomatic asymmetrical pectus with a Haller Index of 10.0 on inspiration, and a cardiac compression index of 5.79. The patient underwent hybrid minimally invasive repair of pectus excavatum followed by bar removal after 3 years. |
| Proband 13 | This is a 42-year-old female with a history of severe symptomatic pectus with a Haller Index of 5.5 and evidence of right heart compression. The patient underwent minimally invasive repair of pectus excavatum followed by bar and bone osteophyte removal after 3 years. |
| Proband 14 | This is a 58-year-old female with a history of severe symptomatic pectus with a Haller Index of 5.4 and evidence of right heart compression. The patient underwent hybrid minimally invasive repair of pectus excavatum followed by bar and bone osteophyte removal after 3 years. |
| Proband 15 | This is a 49-year-old male with a history of severe worsening symptomatic pectus with a Haller Index of 5.67 on inspiration and 9.2 on expiration and evidence of right heart compression. The patient underwent hybrid minimally invasive repair of pectus excavatum followed by bar and bone osteophyte removal after 3 years with no complications. |
| Proband 16 | This is an 18-year-old female with a history of severe symptomatic pectus excavatum with a Haller Index of 4.6 and evidence of right heart compression. The patient underwent minimally invasive repair of pectus excavatum followed by bar and bone osteophyte removal after 3 years with no complications. |
| Family | Gene/Variant | Zygosity | In Silico | ACMG Classification: Criteria Applied |
|---|---|---|---|---|
| PO1 | BMP6 c.379G>A p.Gly127Arg | HET | CADD: 23.8 REVEL: 0.161 | VUS: PM2_supp, BP4 |
| NOTCH3 c.329G>A p.Arg110His | HET | CADD: 22.4 REVEL: 0.580 | VUS: PM2_sup | |
| PO2 | CRTAC1 c.811C>T p.Arg271Trp | HET | CADD: 29.5 REVEL: 0.194 | VUS: PM2_supp, BP4 |
| PO3 | BAZ1B c.2134G>A p.Asp712Asn | HET | CADD: 22.5 REVEL: 0.107 | VUS: PM2_supp, BP4_mod |
| GLG1 c.2935-54T>C p.? | HET | CADD: 5.78 SPLICEAI: AG = 0.33 (AP = −3 bp) | VUS: PM2_supp, PP3 | |
| P14 | BMP6 c.720C>G p.Phe240Leu | HET | CADD: 25.5 REVEL: 0.857 | VUS: PM2_supp, PP3_mod |
| Gene/Variant | Gene Slice | Family | OMIM Disease | Zygosity | In Silico | Conclusions | ACMG Classification: Criteria Applied |
|---|---|---|---|---|---|---|---|
| SMAD4 NM_005359.6:c.-69G>A | TAAD | P02 | Myhre syndrome (MIM: 139210) [AD] Juvenile polyposis/hereditary hemorrhagic telangiectasia syndrome (MIM: 175050) [AD] | proband: HET father: HET mother: REF sister: REF niece: HET | CADD: 5.948 REVEL: NA SPLICEAI: 0.01 | Rare variant that segregates with phenotype. The low CADD suggested a non-deleterious effect. | VUS: PM2_supp, BP7 |
| COL5A1,NM_000093.5:c.-180C>A | TAAD | P04 | Ehlers-Danlos syndrome, classic type, 1 (MIM: 130000) [AD] Fibromuscular dysplasia, multifocal (MIM: 619329) [AD] | proband: HET father: HET mother: REF affected sister: HET unaffected sister: REF | CADD: UNK REVEL: NA SPLCEAI: 0 | Variant segregates with phenotype. The low CADD suggested non-deleterious effect. | B: BS2, BP7 |
| ASXL3, NM_030632.3:c.6236T>C, NP_085135.1:p.Ile2079Thr | Exome-wide candidate list. | P02 | Bainbridge-Ropers syndrome (MIM: 615485) [AD] | proband: HET father: HET mother: REF sister: REF niece: HET | CADD: 25.5 REVEL: 0.29 | This variant segregates with phenotype. Differential strongly indicates that this variant is non-contributory to disease development. | VUS: PM2_supp |
| REST, NM_005612.5:c.70A>G, NP_005603.3:p.Met24Val | Exome-wide candidate list. | P02 | Deafness, autosomal dominant 27 (MIM: 612431) [AD] Fibromatosis, gingival, 5 (MIM: 617626) [AD] | proband: HET father: HET mother: REF sister: REF niece: HET | CADD: 21.1 REVEL: 0.18 | Variant segregates with phenotype (Figure 2A). In silico tools indicate that substitution is tolerated (Figure 2B); however, pathogenic variants in REST are associated with loss-of-function mechanisms. This variant has been associated with pectus deformities in prior studies [14,15] | B: BP4, BS2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farina, J.M.; Olson, R.J.; Dhamija, R.; Bofferding, A.; Sekulic, A.; Egan, J.B.; Jaroszewski, D.E. The Complexity of Familial Inheritance in Pectus Excavatum: A Ten-Family Exome Sequencing Analysis. Genes 2024, 15, 1429. https://doi.org/10.3390/genes15111429
Farina JM, Olson RJ, Dhamija R, Bofferding A, Sekulic A, Egan JB, Jaroszewski DE. The Complexity of Familial Inheritance in Pectus Excavatum: A Ten-Family Exome Sequencing Analysis. Genes. 2024; 15(11):1429. https://doi.org/10.3390/genes15111429
Chicago/Turabian StyleFarina, Juan M., Rory J. Olson, Radhika Dhamija, Anne Bofferding, Aleksandar Sekulic, Jan B. Egan, and Dawn E. Jaroszewski. 2024. "The Complexity of Familial Inheritance in Pectus Excavatum: A Ten-Family Exome Sequencing Analysis" Genes 15, no. 11: 1429. https://doi.org/10.3390/genes15111429
APA StyleFarina, J. M., Olson, R. J., Dhamija, R., Bofferding, A., Sekulic, A., Egan, J. B., & Jaroszewski, D. E. (2024). The Complexity of Familial Inheritance in Pectus Excavatum: A Ten-Family Exome Sequencing Analysis. Genes, 15(11), 1429. https://doi.org/10.3390/genes15111429