
Inference of Genetic Diversity, Population Structure, and Selection Signatures in Xiangxi White Buffalo of China Through Whole-Genome Resequencing
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection and Genome Sequencing
2.3. Genome-Wide Alignment and Variation Detection
2.4. Analysis of Genetic Structure and Relatedness of Buffalo Populations
2.5. Genetic Diversity, Linkage Disequilibrium, and ROH Analysis
2.6. Detection of Selection Signatures
2.7. Enrichment Analyses of Candidate Genes
3. Results
3.1. Whole-Genome Sequencing and SNP Identification
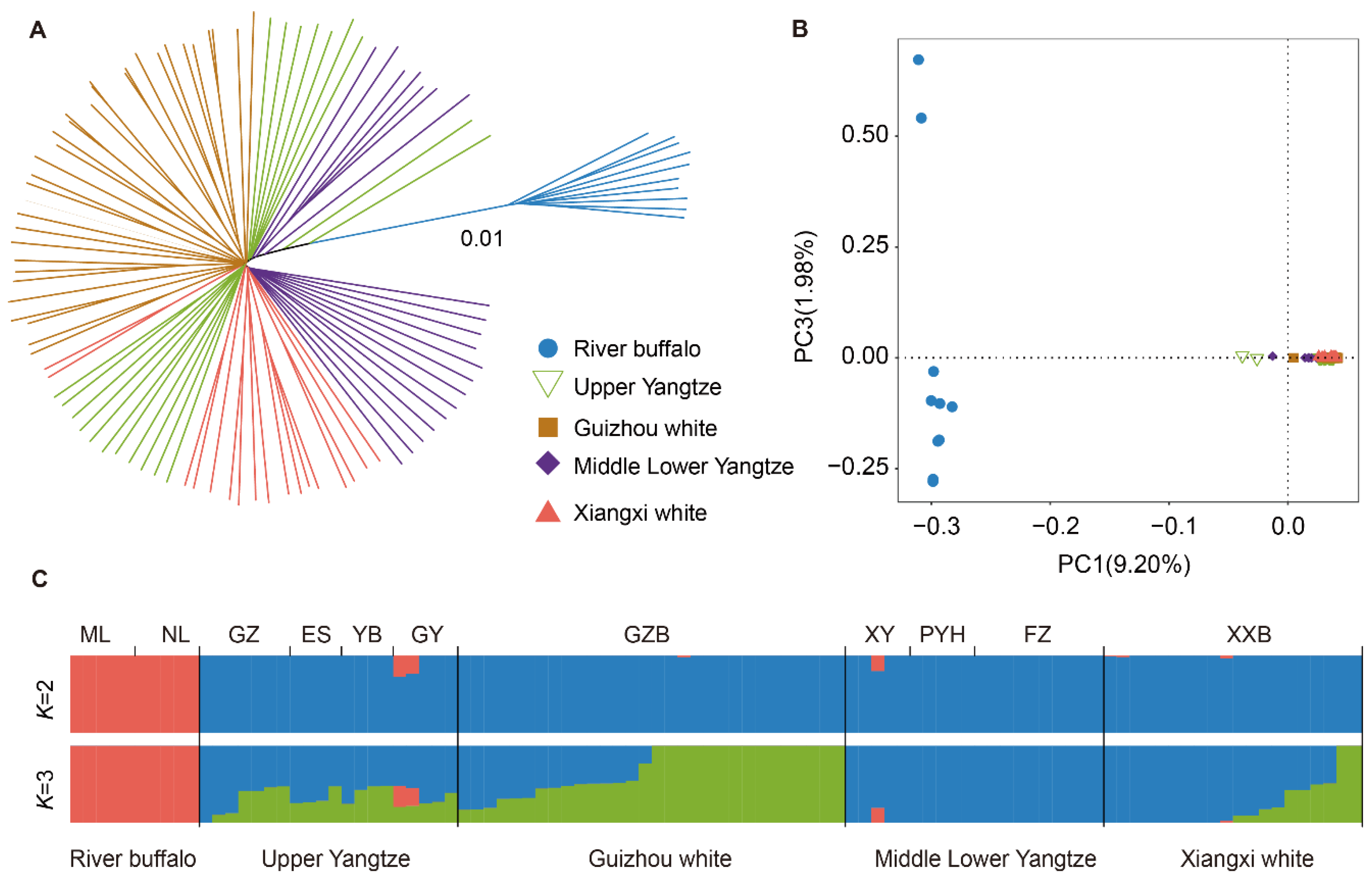
3.2. Population Genetic Structure and Ancestral Relationships
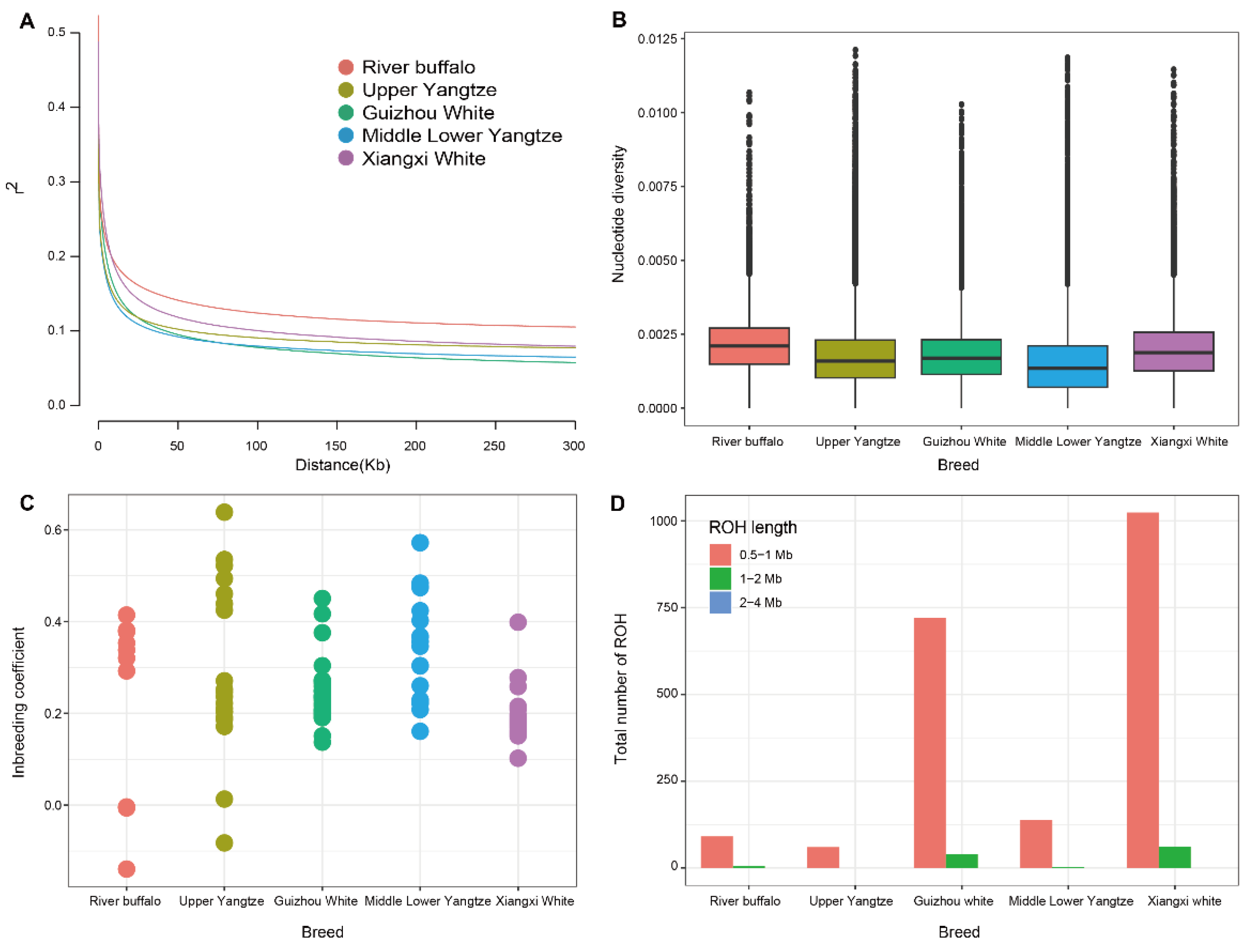
3.3. Patterns of Genomic Variation
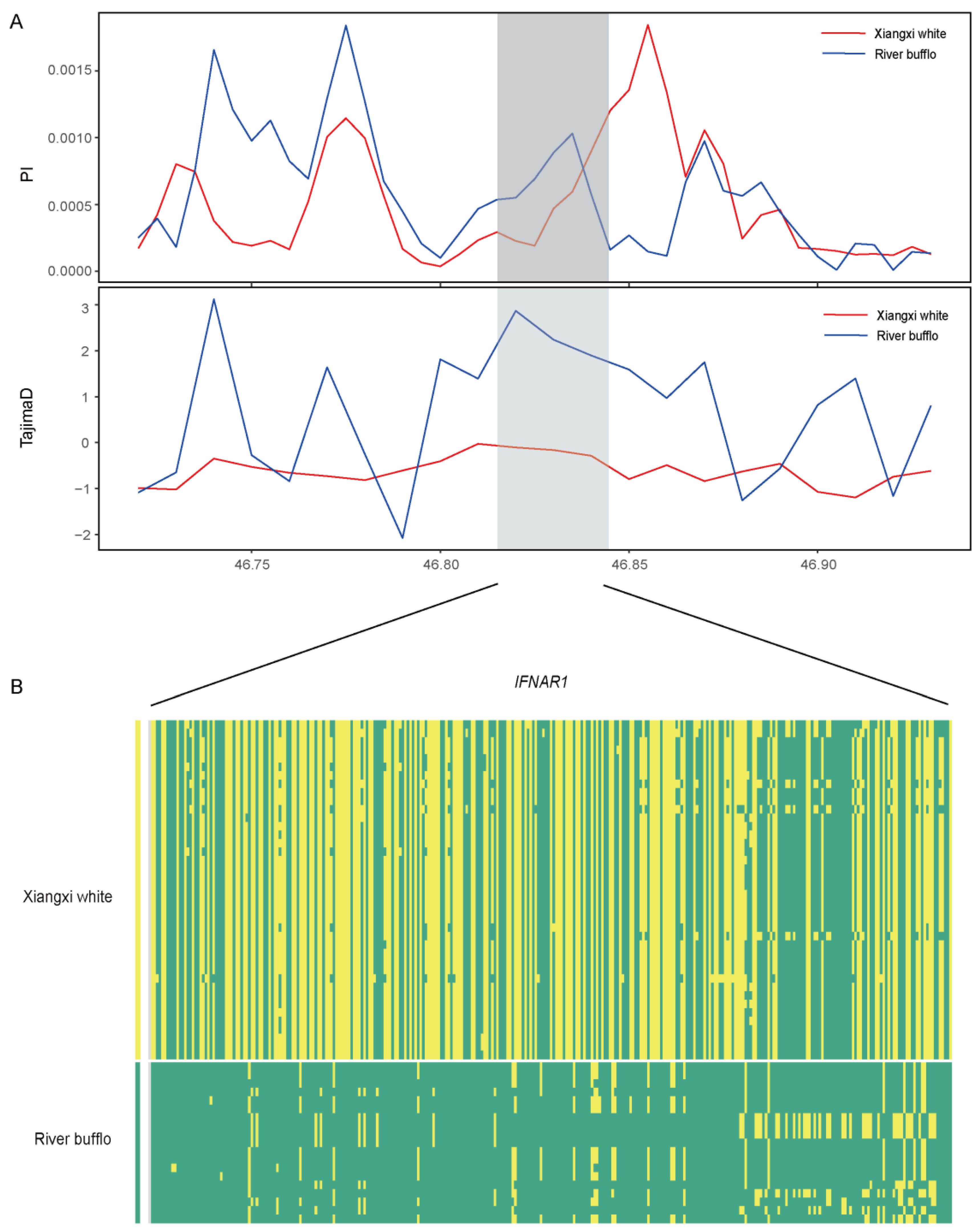
3.4. Patterns of Selection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fischer, H.; Ulbrich, F. Chromosomes of the Murrah buffalo and its crossbreds with the asiatic swamp buffalo (Bubalus bubalis). J. Animal Breed. Genet. 1967, 84, 110–114. [Google Scholar] [CrossRef]
- Windsor, P.; Martin, S.; Khounsy, S.; Young, J.; Thomson, P.; Bush, R. Improved milk production from supplementation of Swamp Buffalo with molasses nutrient blocks containing 10% urea. Dairy 2021, 2, 90–103. [Google Scholar] [CrossRef]
- Michelizzi, V.N.; Dodson, M.V.; Pan, Z.; Amaral, M.E.J.; Michal, J.J.; McLean, D.J.; Womack, J.E.; Jiang, Z. Water buffalo genome science comes of age. Int. J. Biol. Sci. 2010, 6, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Zhao, P.; Si, J.; Fang, L.; Pairo-Castineira, E.; Hu, X.; Xu, Q.; Hou, Y.; Gong, Y.; Liang, Z.; et al. Genomic Analysis Revealed a Convergent Evolution of LINE-1 in Coat Color: A Case Study in Water Buffaloes (Bubalus bubalis). Mol. Biol. Evol. 2021, 38, 1122–1136. [Google Scholar] [CrossRef]
- Kalds, P.; Zhou, S.; Chen, Y.; Wang, X. Modeling animal genomics in mice: An authentic approach for the functional interrogation of evolutionarily and agriculturally critical variants. Anim. Res. One Health 2024, 2, 86–92. [Google Scholar] [CrossRef]
- GB/T 35892-2018; Laboratory Animal—Guideline for Ethical Review of Animal Welfare. Chinese Standard: Beijing, China, 2018.
- Green, M.R.; Sambrook, J. “Molecular Cloning: A Laboratory Manual”, in the Three-Volume Set, 4th ed.; Cold Spring Harbor Laboratory Press: Long Island, NY, USA, 2012. [Google Scholar]
- Leinonen, R.; Sugawara, H.; Shumway, M.; on behalf of the International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows‒Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2013, 2, e190. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Chen, Z.; Zhu, M.; Wu, Q.; Lu, H.; Lei, C.; Ahmed, Z.; Sun, J. Analysis of genetic diversity and selection characteristics using the whole-genome sequencing data of five buffaloes, including Xilin buffalo, in Guangxi, China. Front. Genet. 2023, 13, 1084824. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.S.; Xu, J.Y.; He, W.M.; Yang, T.L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Makanjuola, B.O.; Maltecca, C.; Miglior, F.; Marras, G.; Abdalla, E.A.; Schenkel, F.S.; Baes, C.F. Identification of unique ROH regions with unfavorable effects on production and fertility traits in Canadian Holsteins. Genet. Sel. Evol. 2021, 53, 68. [Google Scholar] [CrossRef]
- Forutan, M.; Ansari, M.S.; Baes, C.; Melzer, N.; Schenkel, F.S.; Sargolzaei, M. Inbreeding and runs of homozygosity before and after genomic selection inNorth American Holstein cattle. BMC Genom. 2018, 19, 98. [Google Scholar] [CrossRef]
- Hudson, R. Estimation of levels of gene flow from DNA sequence data. Genetics 1992, 132, 583. [Google Scholar] [CrossRef]
- Nielsen, R.; Williamson, S.; Kim, Y.; Hubisz, M.J.; Clark, A.G.; Bustamante, C. Genomic scans for selective sweeps using SNP data. Genome Res. 2005, 15, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- DeGiorgio, M.; Huber, C.D.; Hubisz, M.J.; Hellmann, I.; Nielsen, R. SweepFinder2: Increased sensitivity, robustness and flexibility. Bioinformatics 2016, 32, 1895–1897. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 449913–449918. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Szpiech, Z.A.; Hernandez, R.D. Selscan: An efficient multithreaded program to perform EHH-based scans for positive selection. Mol. Biol. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Kong, J.; Qiu, Y.; Yang, X.; Wang, W.; Yan, L. Identification of coregenes and outcomes in hepatocellular carcinoma by bioinformatics analysis. J. Cell Biochem. 2019, 120, 10069–10081. [Google Scholar] [CrossRef]
- El, H.A.; Rocha, D.; Venot, E.; Blanquet, V.; Philippe, R. Long-range linkage disequilibrium in French beef cattle breeds. Genet. Sel. Evol. 2021, 53, 63. [Google Scholar] [CrossRef]
- Wang, W.; Liu, R.; Liang, X.; Zhao, Q.; Qu, P.; Yao, K.; Jiang, M.; Luo, Y.; Zhang, W.; Qing, S. Expression of IFNAR1 and IFNAR2 in cattle placenta during early pregnancy. Reprod. Domest. Anim. 2018, 53, 385–392. [Google Scholar] [CrossRef]
- Doyle, J.L.; Berry, D.P.; Veerkamp, R.F.; Carthy, T.R.; Evans, R.D.; Walsh, S.W.; Purfield, D.C. Genomic regions associated with muscularity in beef cattle differ in five contrasting cattle breeds. Genet. Sel. Evol. 2020, 52, 2. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, P.; He, X.; Liu, Y.; Chu, M. SEMA4G targeted by miR-363-5p regulates the proliferation of granulosa cells in Yunshang black goats. Anim. Res. One Health 2024, 2, 28–38. [Google Scholar] [CrossRef]
- Luo, X.; Li, S.; Liu, Y.; Ahmed, Z.; Wang, F.; Liu, J.; Zhang, J.; Chen, N.; Lei, C.; Huang, B. Assessing the Role of Ancestral Fragments and Selection Signatures by Whole-Genome Scanning in Dehong Humped Cattle at the China–Myanmar Border. Biology 2022, 11, 1331. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lei, H.; Li, J.; Sun, A.; Ahmed, Z.; Duan, H.; Chen, L.; Zhang, B.; Lei, C.; Yi, K. Analysis of genetic diversity and selection signals in Chaling cattle of southern China using whole-genome scan. Anim. Genet. 2023, 54, 284–294. [Google Scholar] [CrossRef]
- Macciotta, N.P.P.; Colli, L.; Cesarani, A.; Ajmone-Marsan, P.; Low, W.Y.; Tearle, R.; Williams, J.L. The distribution of runs of homozygosity in the genome of river and swamp buffaloes reveals a history of adaptation, migration and crossbred events. Genet. Sel. Evol. 2021, 53, 20. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity:windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Shen, J.; Achilli, A.; Chen, N.; Chen, Q.; Dang, R.; Zheng, Z.; Zhang, H.; Zhang, X.; Wang, S.; et al. Genomic analyses reveal distinct genetic architectures and selective pressures in buffaloes. GigaScience 2020, 9, giz166. [Google Scholar] [CrossRef]
- Nuss, P. Anxiety disorders and GABA neurotransmission: A disturbance of modulation. Neuropsychiatr. Dis. Treat. 2015, 11, 165–175. [Google Scholar] [PubMed]
- Palmer, S.; Towne, M.C.; Pearl, P.L.; Pelletier, R.C.; Genetti, C.A.; Shi, J.; Beggs, A.H.; Agrawal, P.B.; Brownstein, C.A. SLC6A1 Mutation and Ketogenic Diet in Epilepsy With Myoclonic-Atonic Seizures. Pediatr. Neurol. 2016, 64, 77–79. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, H. Ca2+-stimulated ADCY1 and ADCY8 regulate distinct aspects of synaptic and cognitive flexibility. Front. Cell Neurosci. 2023, 17, 1215255. [Google Scholar] [CrossRef]
- Cruz, P.M.R.; Hughes, I.; Manzur, A.; Munot, P.; Ramdas, S.; Wright, R.; Breen, C.; Pitt, M.; Pagnamenta, A.T.; Taylor, J.C.; et al. Presynaptic congenital myasthenic syndrome due to three novel mutations in SLC5A7 encoding the sodium-dependant high-affinity choline transporter. Neuromuscul. Disord. 2021, 31, 21–28. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, Y.; Ge, F.; Gao, H.; Zhou, J.; Wu, X.; Qian, C.; Wang, Z.; Wang, Z.; Zhu, B.; et al. Developing a liquid capture chip to accelerate the genetic progress of cattle. Anim. Res. One Health 2024, 2, 204–216. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bian, C.; Luo, Y.; Li, J.; Cheng, H.; He, F.; Duan, H.; Ahmed, Z.; Lei, C.; Yi, K. Inference of Genetic Diversity, Population Structure, and Selection Signatures in Xiangxi White Buffalo of China Through Whole-Genome Resequencing. Genes 2024, 15, 1450. https://doi.org/10.3390/genes15111450
Bian C, Luo Y, Li J, Cheng H, He F, Duan H, Ahmed Z, Lei C, Yi K. Inference of Genetic Diversity, Population Structure, and Selection Signatures in Xiangxi White Buffalo of China Through Whole-Genome Resequencing. Genes. 2024; 15(11):1450. https://doi.org/10.3390/genes15111450
Chicago/Turabian StyleBian, Chenqi, Yang Luo, Jianbo Li, Huan Cheng, Fang He, Hongfeng Duan, Zulfiqar Ahmed, Chuzhao Lei, and Kangle Yi. 2024. "Inference of Genetic Diversity, Population Structure, and Selection Signatures in Xiangxi White Buffalo of China Through Whole-Genome Resequencing" Genes 15, no. 11: 1450. https://doi.org/10.3390/genes15111450
APA StyleBian, C., Luo, Y., Li, J., Cheng, H., He, F., Duan, H., Ahmed, Z., Lei, C., & Yi, K. (2024). Inference of Genetic Diversity, Population Structure, and Selection Signatures in Xiangxi White Buffalo of China Through Whole-Genome Resequencing. Genes, 15(11), 1450. https://doi.org/10.3390/genes15111450