
Genomic Landscape of Chromosome X Factor VIII: From Hemophilia A in Males to Risk Variants in Females

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
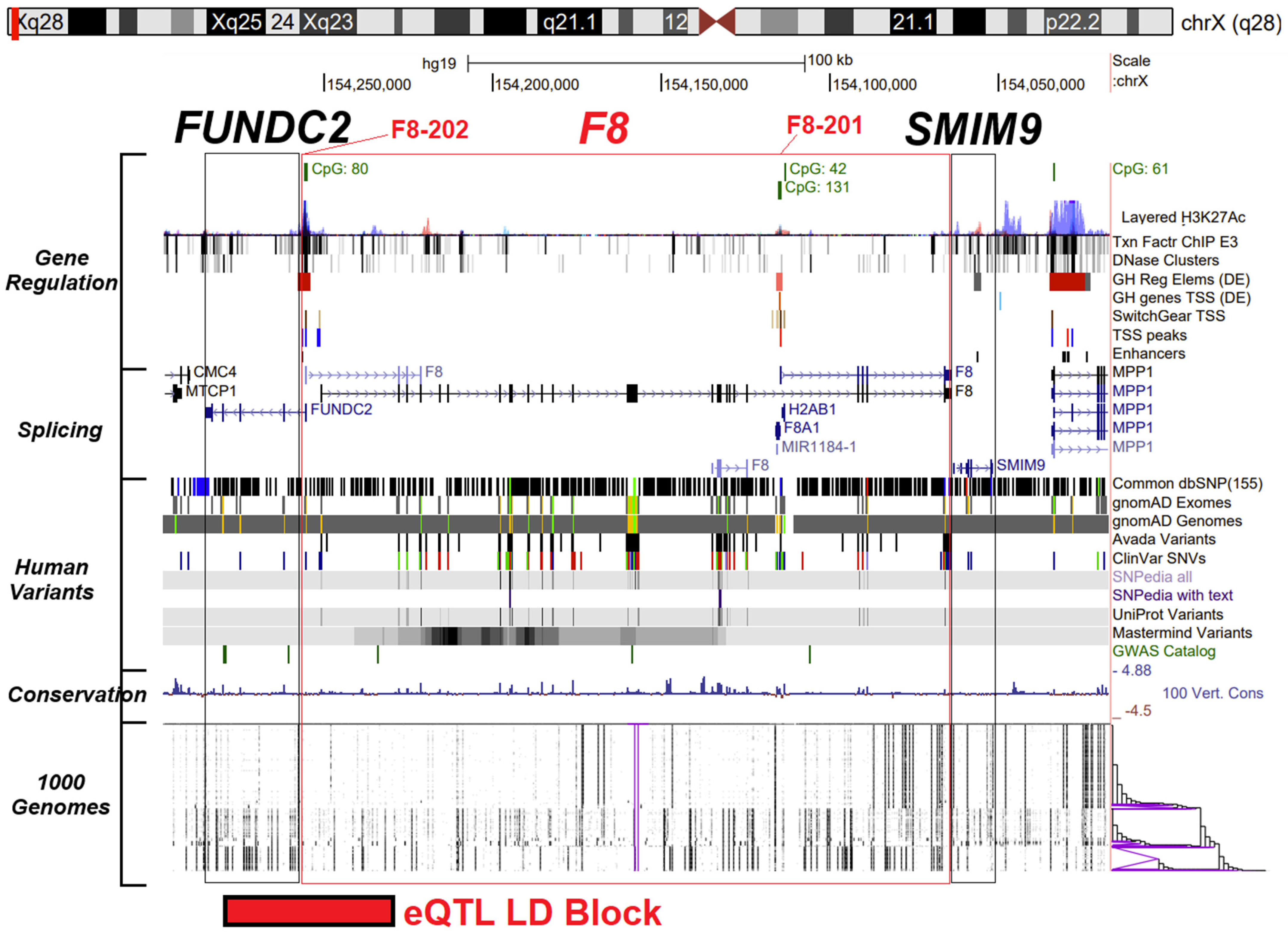
3.1. F8 Gene Promoters, Isoform Expression, and Noncoding Variants
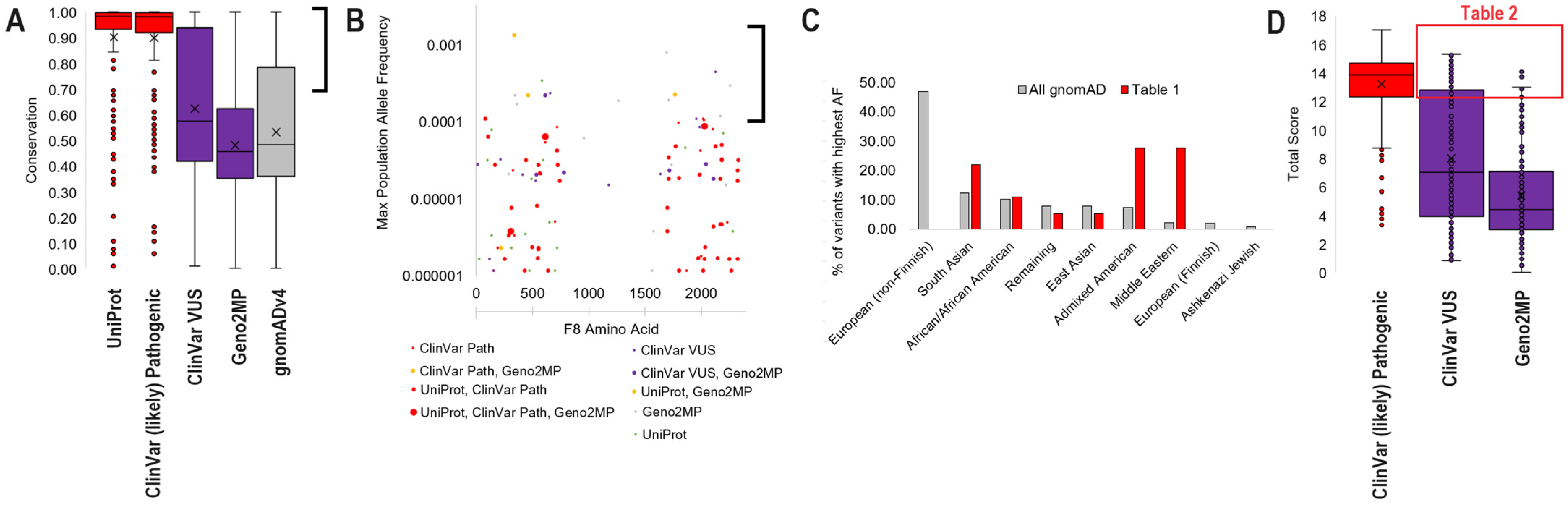
3.2. F8 Protein Conservation and Missense Variant Locations
3.3. Common gnomAD Variants Connected to Phenotype
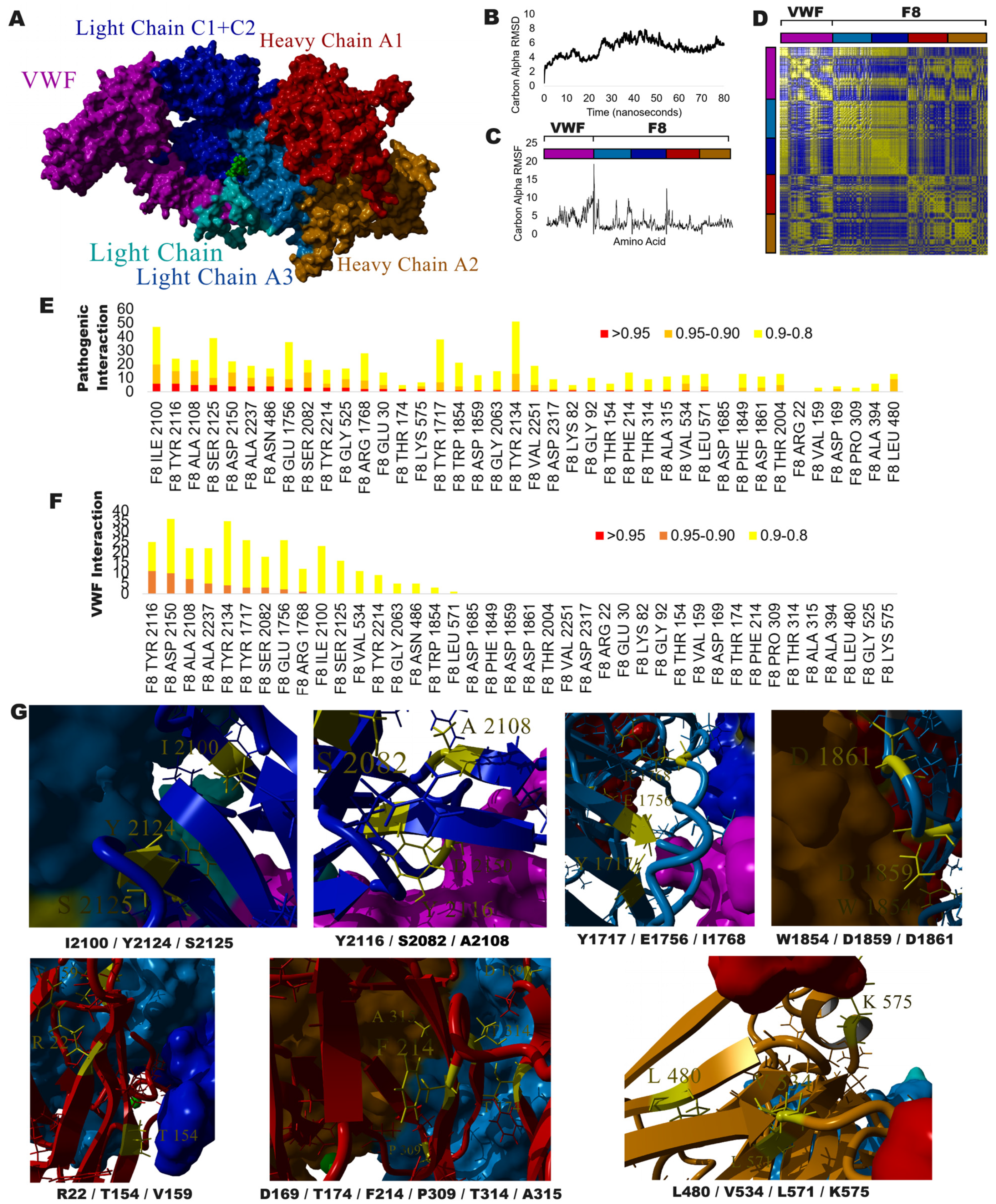
3.4. Exploring VUS with Protein Structure and Molecular Dynamic Simulations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Prokop, J.W.; Tsaih, S.-W.; Faber, A.B.; Boehme, S.; Underwood, A.C.; Troyer, S.; Playl, L.; Milsted, A.; Turner, M.E.; Ely, D.; et al. The Phenotypic Impact of the Male-Specific Region of Chromosome-Y in Inbred Mating: The Role of Genetic Variants and Gene Duplications in Multiple Inbred Rat Strains. Biol. Sex Differ. 2016, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Prokop, J.W.; Deschepper, C.F. Chromosome Y Genetic Variants: Impact in Animal Models and on Human Disease. Physiol. Genom. 2015, 47, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Augui, S.; Nora, E.P.; Heard, E. Regulation of X-Chromosome Inactivation by the X-Inactivation Centre. Nat. Rev. Genet. 2011, 12, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Schwämmle, T.; Schulz, E.G. Regulatory Principles and Mechanisms Governing the Onset of Random X-Chromosome Inactivation. Curr. Opin. Genet. Dev. 2023, 81, 102063. [Google Scholar] [CrossRef]
- Panning, B.; Jaenisch, R. RNA and the Epigenetic Regulation of X Chromosome Inactivation. Cell 1998, 93, 305–308. [Google Scholar] [CrossRef]
- Werner, J.M.; Ballouz, S.; Hover, J.; Gillis, J. Variability of Cross-Tissue X-Chromosome Inactivation Characterizes Timing of Human Embryonic Lineage Specification Events. Dev. Cell 2022, 57, 1995–2008.e5. [Google Scholar] [CrossRef]
- Juchniewicz, P.; Kloska, A.; Portalska, K.; Jakóbkiewicz-Banecka, J.; Węgrzyn, G.; Liss, J.; Głodek, P.; Tukaj, S.; Piotrowska, E. X-Chromosome Inactivation Patterns Depend on Age and Tissue but Not Conception Method in Humans. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2023, 31, 4. [Google Scholar] [CrossRef]
- Shvetsova, E.; Sofronova, A.; Monajemi, R.; Gagalova, K.; Draisma, H.H.M.; White, S.J.; Santen, G.W.E.; Chuva de Sousa Lopes, S.M.; Heijmans, B.T.; van Meurs, J.; et al. Skewed X-Inactivation Is Common in the General Female Population. Eur. J. Hum. Genet. EJHG 2019, 27, 455–465. [Google Scholar] [CrossRef]
- Giovenino, C.; Trajkova, S.; Pavinato, L.; Cardaropoli, S.; Pullano, V.; Ferrero, E.; Sukarova-Angelovska, E.; Carestiato, S.; Salmin, P.; Rinninella, A.; et al. Skewed X-Chromosome Inactivation in Unsolved Neurodevelopmental Disease Cases Can Guide Re-Evaluation For X-Linked Genes. Eur. J. Hum. Genet. EJHG 2023, 31, 1228–1236. [Google Scholar] [CrossRef]
- Berletch, J.B.; Yang, F.; Xu, J.; Carrel, L.; Disteche, C.M. Genes That Escape from X Inactivation. Hum. Genet. 2011, 130, 237–245. [Google Scholar] [CrossRef]
- Fang, H.; Disteche, C.M.; Berletch, J.B. X Inactivation and Escape: Epigenetic and Structural Features. Front. Cell Dev. Biol. 2019, 7, 219. [Google Scholar] [CrossRef] [PubMed]
- Carrel, L.; Willard, H.F. X-Inactivation Profile Reveals Extensive Variability in X-Linked Gene Expression in Females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Tukiainen, T.; Villani, A.-C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; Kirby, A.; et al. Landscape of X Chromosome Inactivation across Human Tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, A.; Brondke, H.; Müsebeck, J.; Pollmann, H.; Srivastava, A.; Oldenburg, J. Molecular Mechanisms Underlying Hemophilia A Phenotype in Seven Females. J. Thromb. Haemost. JTH 2009, 7, 976–982. [Google Scholar] [CrossRef]
- Miller, C.H.; Bean, C.J. Genetic Causes of Haemophilia in Women and Girls. Haemoph. Off. J. World Fed. Hemoph. 2021, 27, e164–e179. [Google Scholar] [CrossRef]
- Renault, N.K.; Dyack, S.; Dobson, M.J.; Costa, T.; Lam, W.L.; Greer, W.L. Heritable Skewed X-Chromosome Inactivation Leads to Haemophilia A Expression in Heterozygous Females. Eur. J. Hum. Genet. EJHG 2007, 15, 628–637. [Google Scholar] [CrossRef]
- Favier, R.; Lavergne, J.M.; Costa, J.M.; Caron, C.; Mazurier, C.; Viémont, M.; Delpech, M.; Valleix, S. Unbalanced X-Chromosome Inactivation with a Novel FVIII Gene Mutation Resulting in Severe Hemophilia A in a Female. Blood 2000, 96, 4373–4375. [Google Scholar] [CrossRef]
- Shinozawa, K.; Niiya, K.; Fujimoto, S.; Bingo, M.; Fukutake, K.; Kinai, E. Trisomy X Conferring Moderate Hemophilia A by Extremely Skewed X-Chromosome Inactivation. Res. Pract. Thromb. Haemost. 2023, 7, 102233. [Google Scholar] [CrossRef]
- Garagiola, I.; Mortarino, M.; Siboni, S.M.; Boscarino, M.; Mancuso, M.E.; Biganzoli, M.; Santagostino, E.; Peyvandi, F. X Chromosome Inactivation: A Modifier of Factor VIII and IX Plasma Levels and Bleeding Phenotype in Haemophilia Carriers. Eur. J. Hum. Genet. EJHG 2021, 29, 241–249. [Google Scholar] [CrossRef]
- Shoukat, H.M.H.; Ghous, G.; Tarar, Z.I.; Shoukat, M.M.; Ajmal, N. Skewed Inactivation of X Chromosome: A Cause of Hemophilia Manifestation in Carrier Females. Cureus 2020, 12, e11216. [Google Scholar] [CrossRef]
- Franchini, M.; Mannucci, P.M. The More Recent History of Hemophilia Treatment. Semin. Thromb. Hemost. 2022, 48, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Jourdy, Y.; Nougier, C.; Roualdes, O.; Fretigny, M.; Durand, B.; Negrier, C.; Vinciguerra, C. Characterization of Five Associations of F8 Missense Mutations Containing FVIII B Domain Mutations. Haemoph. Off. J. World Fed. Hemoph. 2016, 22, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, P.M. Hemophilia Therapy: The Future Has Begun. Haematologica 2020, 105, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-Y.; Koerper, M.A.; Johnson, K.A.; Riske, B.; Baker, J.R.; Ullman, M.; Curtis, R.G.; Poon, J.-L.; Lou, M.; Nichol, M.B. Burden of Illness: Direct and Indirect Costs among Persons with Hemophilia A in the United States. J. Med. Econ. 2015, 18, 457–465. [Google Scholar] [CrossRef]
- Samelson-Jones, B.J.; George, L.A. Adeno-Associated Virus Gene Therapy for Hemophilia. Annu. Rev. Med. 2023, 74, 231–247. [Google Scholar] [CrossRef]
- Boardman, F.K.; Hale, R.; Young, P.J. Newborn Screening for Haemophilia: The Views of Families and Adults Living with Haemophilia in the UK. Haemoph. Off. J. World Fed. Hemoph. 2019, 25, 276–282. [Google Scholar] [CrossRef]
- Peyvandi, F.; Jayandharan, G.; Chandy, M.; Srivastava, A.; Nakaya, S.M.; Johnson, M.J.; Thompson, A.R.; Goodeve, A.; Garagiola, I.; Lavoretano, S.; et al. Genetic Diagnosis of Haemophilia and Other Inherited Bleeding Disorders. Haemoph. Off. J. World Fed. Hemoph. 2006, 12 (Suppl. 3), 82–89. [Google Scholar] [CrossRef]
- Atik, T.; Işık, E.; Onay, H.; Akgün, B.; Shamsali, M.; Kavaklı, K.; Evim, M.; Tüysüz, G.; Özbek, N.Y.; Şahin, F.; et al. Factor 8 Gene Mutation Spectrum of 270 Patients with Hemophilia A: Identification of 36 Novel Mutations. Turk. J. Haematol. Off. J. Turk. Soc. Haematol. 2020, 37, 145–153. [Google Scholar] [CrossRef]
- Gunasekera, D.; Ettinger, R.A.; Nakaya Fletcher, S.; James, E.A.; Liu, M.; Barrett, J.C.; Withycombe, J.; Matthews, D.C.; Epstein, M.S.; Hughes, R.J.; et al. Factor VIII Gene Variants and Inhibitor Risk in African American Hemophilia A Patients. Blood 2015, 126, 895–904. [Google Scholar] [CrossRef]
- Nassar, L.R.; Barber, G.P.; Benet-Pagès, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, B.T.; et al. The UCSC Genome Browser Database: 2023 Update. Nucleic Acids Res. 2023, 51, D1188–D1195. [Google Scholar] [CrossRef]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a Knowledge-Based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium The GTEx Consortium Atlas of Genetic Regulatory Effects across Human Tissues. Science 2020, 369, 1318–1330. [CrossRef] [PubMed]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE Reference Annotation for the Human and Mouse Genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Brown, G.R.; Hem, V.; Katz, K.S.; Ovetsky, M.; Wallin, C.; Ermolaeva, O.; Tolstoy, I.; Tatusova, T.; Pruitt, K.D.; Maglott, D.R.; et al. Gene: A Gene-Centered Information Resource at NCBI. Nucleic Acids Res. 2015, 43, D36–D42. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for Large-Scale Multiple Sequence Alignments. Bioinforma. Oxf. Engl. 2018, 34, 2490–2492. [Google Scholar] [CrossRef]
- Kumar, S.; Tamura, K.; Nei, M. MEGA: Molecular Evolutionary Genetics Analysis Software for Microcomputers. Comput. Appl. Biosci. CABIOS 1994, 10, 189–191. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The Rapid Generation of Mutation Data Matrices from Protein Sequences. Comput. Appl. Biosci. CABIOS 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Prokop, J.W.; Jdanov, V.; Savage, L.; Morris, M.; Lamb, N.; VanSickle, E.; Stenger, C.L.; Rajasekaran, S.; Bupp, C.P. Computational and Experimental Analysis of Genetic Variants. Compr. Physiol. 2022, 12, 3303–3336. [Google Scholar] [CrossRef]
- Prokop, J.W.; Lazar, J.; Crapitto, G.; Smith, D.C.; Worthey, E.A.; Jacob, H.J. Molecular Modeling in the Age of Clinical Genomics, the Enterprise of the next Generation. J. Mol. Model. 2017, 23, 75. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public Archive of Interpretations of Clinically Relevant Variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.J.; Shendure, J.A.; Valle, D.; Hamosh, A.; Lupski, J.R.; Gibbs, R.A.; Boerwinkle, E.; Lifton, R.P.; Gerstein, M.; Gunel, M.; et al. The Centers for Mendelian Genomics: A New Large-Scale Initiative to Identify the Genes Underlying Rare Mendelian Conditions. Am. J. Med. Genet. A. 2012, 158A, 1523–1525. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Francioli, L.C.; Goodrich, J.K.; Collins, R.L.; Kanai, M.; Wang, Q.; Alföldi, J.; Watts, N.A.; Vittal, C.; Gauthier, L.D.; et al. A Genomic Mutational Constraint Map Using Variation in 76,156 Human Genomes. Nature 2024, 625, 92–100. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the Deleteriousness of Variants throughout the Human Genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. Editor. Board Jonathan Haines Al 2013, 07, Unit7.20. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the Precision of Comparative Models with YASARA NOVA—A Self-Parameterizing Force Field. Proteins 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H.; Markley, J.L. The Worldwide Protein Data Bank (wwPDB): Ensuring a Single, Uniform Archive of PDB Data. Nucleic Acids Res. 2007, 35, D301–D303. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Hinds, D.A.; Buil, A.; Ziemek, D.; Martinez-Perez, A.; Malik, R.; Folkersen, L.; Germain, M.; Mälarstig, A.; Brown, A.; Soria, J.M.; et al. Genome-Wide Association Analysis of Self-Reported Events in 6135 Individuals and 252 827 Controls Identifies 8 Loci Associated with Thrombosis. Hum. Mol. Genet. 2016, 25, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of Functional Variation in Personal Genomes Using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- Kapteijn, M.Y.; Lanting, V.R.; Kaptein, F.H.J.; Guman, N.A.M.; Laghmani, E.H.; Kuipers, T.B.; Mei, H.; Goeman, J.J.; Mulder, F.I.; van Duinen, S.G.; et al. RNA-Sequencing to Discover Genes and Signaling Pathways Associated with Venous Thromboembolism in Glioblastoma Patients: A Case-Control Study. Thromb. Res. 2023, 232, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Valleix, S.; Jeanny, J.C.; Elsevier, S.; Joshi, R.L.; Fayet, P.; Bucchini, D.; Delpech, M. Expression of Human F8B, a Gene Nested within the Coagulation Factor VIII Gene, Produces Multiple Eye Defects and Developmental Alterations in Chimeric and Transgenic Mice. Hum. Mol. Genet. 1999, 8, 1291–1301. [Google Scholar] [CrossRef]
- Sanders, M.; Lawlor, J.M.J.; Li, X.; Schuen, J.N.; Millard, S.L.; Zhang, X.; Buck, L.; Grysko, B.; Uhl, K.L.; Hinds, D.; et al. Genomic, Transcriptomic, and Protein Landscape Profile of CFTR and Cystic Fibrosis. Hum. Genet. 2021, 140, 423–439. [Google Scholar] [CrossRef]
- Charron, J.G.; Hernandez, A.; Bilinovich, S.M.; Vogt, D.L.; Bedinger, L.A.; Seaver, L.H.; Williams, M.; Devries, S.; Campbell, D.B.; Bupp, C.P.; et al. N-Methyl-d-Aspartate (NMDA) Receptor Genetics: The Power of Paralog Homology and Protein Dynamics in Defining Dominant Genetic Variants. Am. J. Med. Genet. A. 2022, 188, 556–568. [Google Scholar] [CrossRef]
- Soucie, J.M.; Miller, C.H.; Dupervil, B.; Le, B.; Buckner, T.W. Occurrence Rates of Haemophilia among Males in the United States Based on Surveillance Conducted in Specialized Haemophilia Treatment Centres. Haemoph. Off. J. World Fed. Hemoph. 2020, 26, 487–493. [Google Scholar] [CrossRef]
- DiMichele, D.M. Navigating Speed Bumps on the Innovation Highway in Hemophilia Therapeutics. HemaSphere 2018, 2, e144. [Google Scholar] [CrossRef]
- Uster, D.W.; Chowdary, P.; Riddell, A.; Garcia, C.; Aradom, E.; Musarara, M.; Wicha, S.G. Dosing for Personalized Prophylaxis in Hemophilia A Highly Varies on the Underlying Population Pharmacokinetic Models. Ther. Drug Monit. 2022, 44, 665–673. [Google Scholar] [CrossRef]
- Henderson, M.L.; Zieba, J.K.; Li, X.; Campbell, D.B.; Williams, M.R.; Vogt, D.L.; Bupp, C.P.; Edgerly, Y.M.; Rajasekaran, S.; Hartog, N.L.; et al. Gene Therapy for Genetic Syndromes: Understanding the Current State to Guide Future Care. Biotech Basel Switz. 2024, 13, 1. [Google Scholar] [CrossRef]
- Plug, I.; Mauser-Bunschoten, E.P.; Bröcker-Vriends, A.H.J.T.; van Amstel, H.K.P.; van der Bom, J.G.; van Diemen-Homan, J.E.M.; Willemse, J.; Rosendaal, F.R. Bleeding in Carriers of Hemophilia. Blood 2006, 108, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Ay, C.; Thom, K.; Abu-Hamdeh, F.; Horvath, B.; Quehenberger, P.; Male, C.; Mannhalter, C.; Pabinger, I. Determinants of Factor VIII Plasma Levels in Carriers of Haemophilia A and in Control Women. Haemoph. Off. J. World Fed. Hemoph. 2010, 16, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Candy, V.; Whitworth, H.; Grabell, J.; Thibeault, L.; Harpell, L.; Bowman, M.; Good, D.; Hopman, W.M.; Sidonio, R.F.; James, P.D. A Decreased and Less Sustained Desmopressin Response in Hemophilia A Carriers Contributes to Bleeding. Blood Adv. 2018, 2, 2629–2636. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| gnomAD | gnomAD | gnomAD | gnomAD | gnomAD | Geno2MP | ClinVar and Geno2MP | |||
|---|---|---|---|---|---|---|---|---|---|
| Variant | Conservation | Group | Homozygote | Hemizygote | Overall AF | Max AF | Population | Homozygous | Phenotypes |
| E340K | 0.96 | UniProt, Geno2MP | 0 | 45 | 7.61E-05 | 1.39E-03 | South Asian | 2 | Nephrotic syndrome, Abnormal muscle physiology |
| E1690G | 0.87 | Geno2MP | 0 | 2 | 2.37E-05 | 8.28E-04 | East Asian | 0 | Bladder exstrophy |
| S2125T | 0.96 | ClinVar VUS | 0 | 14 | 3.73E-05 | 4.58E-04 | Middle Eastern | 0 | Hereditary factor VIII deficiency disease |
| N583S | 0.99 | UniProt | 1 | 17 | 1.82E-05 | 3.52E-04 | South Asian | 0 | - |
| T2256S | 0.86 | Geno2MP | 0 | 3 | 1.16E-05 | 3.06E-04 | Admixed American | 1 | Fatigable weakness |
| Y657H | 0.94 | ClinVar VUS | 0 | 3 | 6.38E-06 | 2.42E-04 | Middle Eastern | 0 | Hereditary factor VIII deficiency disease |
| Q1764R | 0.89 | ClinVar Path, Geno2MP | 0 | 56 | 1.32E-04 | 2.31E-04 | Remaining | 1 | Thrombophilia, Heterotaxy |
| R458H | 0.93 | ClinVar Path, Geno2MP | 0 | 20 | 4.96E-05 | 2.29E-04 | Middle Eastern | 0 | Abnormality of hindbrain morphology |
| R612H | 0.91 | ClinVar VUS, Geno2MP | 0 | 7 | 2.48E-05 | 2.29E-04 | Middle Eastern | 1 | Aplasia cutis congenita, Hereditary factor VIII deficiency disease |
| K344Q | 0.77 | Geno2MP | 0 | 4 | 9.92E-06 | 2.28E-04 | Middle Eastern | 0 | Ebstein’s anomaly of the tricuspid valve |
| P1707L | 0.82 | Geno2MP | 0 | 19 | 4.88E-05 | 1.98E-04 | Admixed American | 3 | Retinal degeneration |
| P1265Q | 0.75 | Geno2MP | 0 | 6 | 1.24E-05 | 1.94E-04 | South Asian | 0 | Nephrotic syndrome |
| D364N | 0.74 | Geno2MP | 0 | 7 | 1.16E-05 | 1.75E-04 | Admixed American | 0 | Progressive muscle weakness |
| R2166Q | 0.92 | Geno2MP | 0 | 15 | 4.64E-05 | 1.23E-04 | South Asian | 2 | Abnormality of limbs |
| P83R | 0.83 | UniProt, ClinVar Path | 0 | 3 | 8.22E-06 | 1.14E-04 | Admixed American | 0 | Hereditary factor VIII deficiency disease |
| R2016Q | 0.95 | ClinVar Path | 0 | 3 | 8.20E-06 | 1.14E-04 | African/African American | 0 | Hereditary factor VIII deficiency disease |
| Q1955E | 0.92 | ClinVar VUS | 0 | 0 | 2.73E-06 | 1.14E-04 | African/African American | 0 | Hereditary factor VIII deficiency disease |
| E2023K | 0.95 | ClinVar Path | 0 | 4 | 1.16E-05 | 1.09E-04 | Admixed American | 0 | Thrombophilia |
| Variant | Group | 322 Species Conservation | Total Score | gnomAD Allele Count | Geno2MP Homozygous | gnomAD Homozygote | CADD_PHRED | SIFT | PolyPhen |
|---|---|---|---|---|---|---|---|---|---|
| P309L | ClinVar VUS | 0.95 | 15.31 | 0 | 0 | 0 | 26.2 | deleterious | probably_damaging |
| W1854C | ClinVar VUS | 1.00 | 15.23 | 0 | 0 | 0 | 26.4 | deleterious | probably_damaging |
| Y2134H | ClinVar VUS | 1.00 | 15.00 | 0 | 0 | 0 | 26.1 | deleterious | probably_damaging |
| Y2116H | ClinVar VUS | 0.99 | 14.99 | 0 | 0 | 0 | 26.8 | deleterious | probably_damaging |
| D2150V | ClinVar VUS | 0.99 | 14.92 | 0 | 0 | 0 | 28.6 | deleterious | probably_damaging |
| D169N | ClinVar VUS | 1.00 | 14.85 | 0 | 0 | 0 | 25.6 | deleterious | probably_damaging |
| T174I | ClinVar VUS | 1.00 | 14.77 | 0 | 0 | 0 | 24.7 | deleterious | probably_damaging |
| G92S | ClinVar VUS | 1.00 | 14.76 | 0 | 0 | 0 | 24.8 | deleterious | probably_damaging |
| A2237S | ClinVar VUS | 0.99 | 14.55 | 0 | 0 | 0 | 24.9 | deleterious | probably_damaging |
| L571R | ClinVar VUS | 1.00 | 14.50 | 0 | 0 | 0 | 26.4 | deleterious | probably_damaging |
| T2004A | ClinVar VUS | 1.00 | 14.45 | 0 | 0 | 0 | 25.7 | deleterious | probably_damaging |
| D1861G | ClinVar VUS | 0.87 | 14.43 | 0 | 0 | 0 | 26.1 | deleterious | probably_damaging |
| N486I | ClinVar VUS | 0.99 | 14.24 | 0 | 0 | 0 | 26.1 | deleterious | probably_damaging |
| V2251M | ClinVar VUS | 0.98 | 14.18 | 0 | 0 | 0 | 24.6 | deleterious | probably_damaging |
| T314P | ClinVar VUS | 0.99 | 14.17 | 0 | 0 | 0 | 25.2 | deleterious | probably_damaging |
| I2100T | ClinVar VUS | 0.89 | 14.13 | 0 | 0 | 0 | 24.4 | deleterious | probably_damaging |
| G525R | ClinVar VUS | 0.92 | 14.12 | 4 | 0 | 0 | 25.1 | deleterious | probably_damaging |
| G2063E | Geno2MP | 0.98 | 14.07 | 0 | 0 | 0 | 28.8 | deleterious | probably_damaging |
| L480P | ClinVar VUS | 0.61 | 14.06 | 0 | 0 | 0 | 25.7 | deleterious | probably_damaging |
| E1756V | ClinVar VUS | 0.95 | 13.97 | 0 | 0 | 0 | 27.4 | deleterious | probably_damaging |
| S2125T | ClinVar VUS | 0.96 | 13.94 | 45 | 0 | 0 | 24.7 | deleterious | probably_damaging |
| A2108P | ClinVar VUS | 0.99 | 13.85 | 0 | 0 | 0 | 24.2 | deleterious | probably_damaging |
| R1768S | Geno2MP | 0.98 | 13.71 | 0 | 0 | 0 | 25.6 | deleterious | probably_damaging |
| Y1717H | ClinVar VUS, Geno2MP | 1.00 | 13.65 | 3 | 0 | 0 | 25.8 | deleterious | probably_damaging |
| R22K | ClinVar VUS | 0.98 | 13.43 | 0 | 0 | 0 | 25.9 | deleterious | probably_damaging |
| F1849I | ClinVar VUS | 0.98 | 13.43 | 0 | 0 | 0 | 26 | deleterious | probably_damaging |
| E30G | ClinVar VUS | 0.96 | 13.35 | 0 | 0 | 0 | 23.7 | deleterious | probably_damaging |
| K82E | ClinVar VUS | 0.98 | 13.28 | 0 | 0 | 0 | 24.8 | deleterious | probably_damaging |
| A394D | ClinVar VUS | 0.97 | 13.27 | 0 | 0 | 0 | 25.2 | deleterious | probably_damaging |
| D1859V | ClinVar VUS | 0.58 | 13.24 | 0 | 0 | 0 | 26 | deleterious | probably_damaging |
| K575T | ClinVar VUS | 0.98 | 13.13 | 1 | 0 | 0 | 24.6 | deleterious | probably_damaging |
| T154I | ClinVar VUS | 0.96 | 13.05 | 1 | 0 | 0 | 24.3 | deleterious | probably_damaging |
| S2082N | ClinVar VUS, UniProt | 0.97 | 13.03 | 0 | 0 | 0 | 24.6 | deleterious | probably_damaging |
| A315G | ClinVar VUS | 0.98 | 13.01 | 0 | 0 | 0 | 24.6 | deleterious | probably_damaging |
| Y2214C | Geno2MP | 0.70 | 12.98 | 7 | 0 | 0 | 25.6 | deleterious | probably_damaging |
| D1685G | Geno2MP | 0.70 | 12.86 | 0 | 2 | 0 | 25.2 | deleterious | probably_damaging |
| V534A | ClinVar VUS, Geno2MP | 0.74 | 12.80 | 2 | 1 | 0 | 25.6 | deleterious | probably_damaging |
| A2108V | ClinVar VUS, Geno2MP | 0.99 | 12.56 | 1 | 1 | 0 | 25.4 | deleterious | possibly_damaging |
| D2317A | Geno2MP | 0.62 | 12.50 | 3 | 1 | 0 | 25.2 | deleterious | probably_damaging |
| F214Y | ClinVar VUS | 0.92 | 12.49 | 1 | 0 | 0 | 24.9 | deleterious | probably_damaging |
| V159A | ClinVar VUS, UniProt | 0.52 | 12.33 | 0 | 0 | 0 | 24.9 | deleterious | possibly_damaging |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morris, O.; Morris, M.; Jobe, S.; Bhargava, D.; Krueger, J.M.; Arora, S.; Prokop, J.W.; Stenger, C. Genomic Landscape of Chromosome X Factor VIII: From Hemophilia A in Males to Risk Variants in Females. Genes 2024, 15, 1522. https://doi.org/10.3390/genes15121522
Morris O, Morris M, Jobe S, Bhargava D, Krueger JM, Arora S, Prokop JW, Stenger C. Genomic Landscape of Chromosome X Factor VIII: From Hemophilia A in Males to Risk Variants in Females. Genes. 2024; 15(12):1522. https://doi.org/10.3390/genes15121522
Chicago/Turabian StyleMorris, Olivia, Michele Morris, Shawn Jobe, Disha Bhargava, Jena M. Krueger, Sanjana Arora, Jeremy W. Prokop, and Cynthia Stenger. 2024. "Genomic Landscape of Chromosome X Factor VIII: From Hemophilia A in Males to Risk Variants in Females" Genes 15, no. 12: 1522. https://doi.org/10.3390/genes15121522
APA StyleMorris, O., Morris, M., Jobe, S., Bhargava, D., Krueger, J. M., Arora, S., Prokop, J. W., & Stenger, C. (2024). Genomic Landscape of Chromosome X Factor VIII: From Hemophilia A in Males to Risk Variants in Females. Genes, 15(12), 1522. https://doi.org/10.3390/genes15121522