
The Newborn Screening Programme Revisited: An Expert Opinion on the Challenges of Rett Syndrome

{kind=link}
Abstract
1. Background
Rett Syndrome
2. The Gene Causing the Condition Can Be Reliably Detected
2.1. Genetic Prognosticators of RTT
2.2. Genetic Modifiers
3. The Rare Condition Would Have a Harmful Impact on the Child If Undiagnosed
The Burden of RTT
4. Early or Presymptomatic Intervention Would Improve the Child’s Quality of Life
Early Presymptomatic Testing
5. Early Intervention Is Equitable and Accessible to All
5.1. Early Intervention
5.2. Transformative Therapies
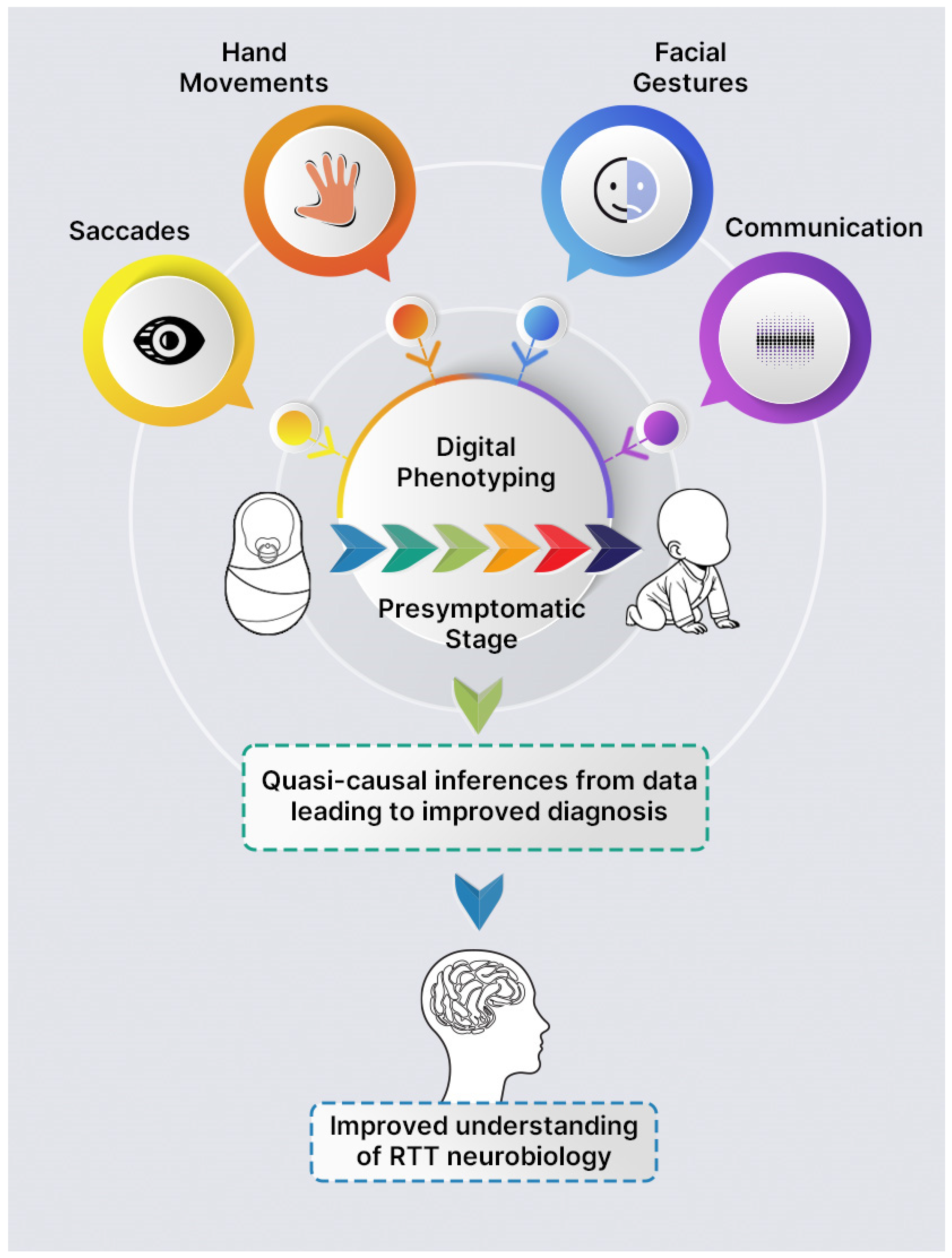
6. Possible Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stark, Z.; Scott, R.H. Genomic newborn screening for rare diseases. Nat. Rev. Genet. 2023, 24, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Stark, Z.; Dolman, L.; Manolio, T.A.; Ozenberger, B.; Hill, S.L.; Caulfied, M.J.; Levy, Y.; Glazer, D.; Wilson, J.; Lawler, M.; et al. Integrating Genomics into Healthcare: A Global Responsibility. Am. J. Hum. Genet. 2019, 104, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, C.; Firth, H.V.; Wilkie, A.O.M.; Newman, W.; Raymond, F.L.; Tomlinson, I.; Lachmann, R.; Wright, C.F.; Wordsworth, S.; George, A.; et al. Population screening requires robust evidence—Genomics is no exception. Lancet 2024, 403, 583–586. [Google Scholar] [CrossRef]
- Jiang, S.; Wang, H.; Gu, Y. Genome Sequencing for Newborn Screening—An Effective Approach for Tackling Rare Diseases. JAMA Netw. Open 2023, 6, e2331141. [Google Scholar] [CrossRef] [PubMed]
- Green, R.C.; Shah, N.; Genetti, C.A.; Yu, T.; Zettler, B.; Uveges, M.K.; Ceyhan-Birsoy, O.; Lebo, M.S.; Pereira, S.; Agrawal, P.B.; et al. Actionability of unanticipated monogenic disease risks in newborn genomic screening: Findings from the BabySeq Project. Am. J. Hum. Genet. 2023, 110, 1034–1045. [Google Scholar] [CrossRef]
- Knoppers, B.M.; Bonilha, A.E.; Laberge, A.-M.; Ahmed, A.; Newson, A.J. Genomic sequencing in newborn screening: Balancing consent with the right of the asymptomatic at-risk child to be found. Eur. J. Hum. Genet. 2024. [Google Scholar] [CrossRef]
- ICoNS. What if We Could Prevent Treatable Disease Before it Developed? Available online: https://www.iconseq.org/ (accessed on 27 November 2024).
- Genomics. Newborn Genomes Programme. Available online: https://www.genomicsengland.co.uk/initiatives/newborns (accessed on 25 October 2024).
- NHS. Newborn Blood Spot Test. Available online: https://www.nhs.uk/conditions/baby/newborn-screening/blood-spot-test/ (accessed on 25 October 2024).
- Genomics. Conditions List. Available online: https://www.genomicsengland.co.uk/initiatives/newborns/choosing-conditions/conditions-list-generation-study (accessed on 25 October 2024).
- Genomics. How We Choose Conditions. Available online: https://www.genomicsengland.co.uk/initiatives/newborns/choosing-conditions (accessed on 25 October 2024).
- Palmieri, M.; Pozzer, D.; Landsberger, N. Advanced genetic therapies for the treatment of Rett syndrome: State of the art and future perspectives. Front. Neurosci. 2023, 17, 1172805. [Google Scholar] [CrossRef]
- Singh, J.; Lanzarini, E.; Santosh, P. Autonomic dysfunction and sudden death in patients with Rett syndrome: A systematic review. J. Psychiatry Neurosci. 2020, 45, 150–181. [Google Scholar] [CrossRef]
- Vilvarajan, S.; McDonald, M.; Douglas, L.; Newham, J.; Kirkland, R.; Tzannes, G.; Tay, D.; Christodoulou, J.; Thompson, S.; Ellaway, C. Multidisciplinary Management of Rett Syndrome: Twenty Years’ Experience. Genes 2023, 14, 1607. [Google Scholar] [CrossRef]
- May, D.; Kponee-Shovein, K.; Neul, J.L.; Percy, A.K.; Mahendran, M.; Downes, N.; Chen, G.; Watson, T.; Pichard, D.C.; Kennedy, M.; et al. Characterizing the journey of Rett syndrome among females in the United States: A real-world evidence study using the Rett syndrome natural history study database. J. Neurodev. Disord. 2024, 16, 42. [Google Scholar] [CrossRef]
- Zade, K.; Campbell, C.; Bach, S.; Fernandes, H.; Tropea, D. Rett syndrome in Ireland: A demographic study. Orphanet J. Rare Dis. 2024, 19, 34. [Google Scholar] [CrossRef] [PubMed]
- Neul, J.L.; Kaufmann, W.E.; Glaze, D.G.; Christodoulou, J.; Clarke, A.J.; Bahi-Buisson, N.; Leonard, H.; Bailey, M.E.; Schanen, N.C.; Zappella, M.; et al. RettSearch Consortium. Rett syndrome: Revised diagnostic criteria and nomenclature. Ann. Neurol. 2010, 68, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007, 315, 1143–1147. [Google Scholar] [CrossRef]
- Suter, B.; Treadwell-Deering, D.; Zoghbi, H.Y.; Glaze, D.G.; Neul, J.L. Brief report: MECP2 mutations in people without Rett syndrome. J. Autism Dev. Disord. 2014, 44, 703–711. [Google Scholar] [CrossRef]
- Halbach, N.S.; Smeets, E.E.; van den Braak, N.; van Roozendaal, K.E.; Blok, R.M.; Schrander-Stumpel, C.T.; Frijns, J.P.; Maaskant, M.A.; Curfs, L.M. Genotype-phenotype relationships as prognosticators in Rett syndrome should be handled with care in clinical practice. Am. J. Med. Genet. A 2012, 158, 340–350. [Google Scholar] [CrossRef]
- Canton, A.P.M.; Tinano, F.R.; Guasti, L.; Montenegro, L.R.; Ryan, F.; Shears, D.; de Melo, M.E.; Gomes, L.G.; Piana, M.P.; Brauner, R.; et al. Rare variants in the MECP2 gene in girls with central precocious puberty: A translational cohort study. Lancet Diabetes Endocrinol. 2023, 11, 545–554. [Google Scholar] [CrossRef]
- Ziegler, A.; Chung, W.K. Universal newborn screening using genome sequencing: Early experience from the GUARDIAN study. Pediatr. Res. 2024. [Google Scholar] [CrossRef]
- van den Heuvel, L.M.; van der Pal, S.M.; Verschoof-Puite, R.K.; Klapwijk, J.E.; Elsinghorst, E.; Dekkers, E.; van der Ploeg, C.P.; Henneman, L. Psychosocial Impact of a True-Positive, False-Positive, or Inconclusive Newborn Bloodspot Screening Result: A Questionnaire Study among Parents. Int. J. Neonatal Screen. 2024, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A.N.; Gallagher, R.C.; Wang, Y.; Currier, R.J.; Amatuni, G.; Bassaganyas, L.; Chen, F.; Kundu, K.; Kvale, M.; Mooney, S.D.; et al. The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat. Med. 2020, 26, 1392–1397. [Google Scholar] [CrossRef]
- Fatumo, S.; Chikowore, T.; Choudhury, A.; Ayub, M.; Martin, A.R.; Kuchenbaecker, K. A roadmap to increase diversity in genomic studies. Nat. Med. 2022, 28, 243–250. [Google Scholar] [CrossRef]
- Boycott, K.M.; Innes, A.M. When One Diagnosis Is Not Enough. N. Engl. J. Med. 2017, 376, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, X.; Chen, Z.; He, R.; Zhang, Y.; Dong, H.; Ma, Y.; Wu, T.; Wang, Q.; Ding, Y.; et al. Dual rare genetic diseases in five pediatric patients: Insights from next-generation diagnostic methods. Orphanet J. Rare Dis. 2024, 19, 159. [Google Scholar] [CrossRef] [PubMed]
- Hannah-Shmouni, F.; Al-Shahoumi, R.; Brady, L.I.; Wu, L.; Frei, J.; Tarnopolsky, M.A. Dual molecular diagnoses in a neurometabolic specialty clinic. Am. J. Med. Genet. Part A 2021, 185, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Akdemir, Z.H.C.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Wilkins, G.; Goodman-Vincent, E.; Chishti, S.; Bonilla Guerrero, R.; Fiori, F.; Ameenpur, S.; McFadden, L.; Zahavi, Z.; Santosh, P. Using Precision Medicine to Disentangle Genotype-Phenotype Relationships in Twins with Rett Syndrome: A Case Report. Curr. Issues Mol. Biol. 2024, 46, 8424–8440. [Google Scholar] [CrossRef]
- Singh, J.; Wilkins, G.; Goodman-Vincent, E.; Chishti, S.; Bonilla Guerrero, R.; McFadden, L.; Zahavi, Z.; Santosh, P. Co-Occurring Methylenetetrahydrofolate Reductase (MTHFR) rs1801133 and rs1801131 Genotypes as Associative Genetic Modifiers of Clinical Severity in Rett Syndrome. Brain Sci. 2024, 14, 624. [Google Scholar] [CrossRef]
- Artuso, R.; Papa, F.T.; Grillo, E.; Mucciolo, M.; Yasui, D.H.; Dunaway, K.W.; Disciglio, V.; Mencarelli, M.A.; Pollazzon, M.; Zappella, M.; et al. Investigation of modifier genes within copy number variations in Rett syndrome. J. Hum. Genet. 2011, 56, 508–515. [Google Scholar] [CrossRef]
- Genomics. How We Choose Conditions. Available online: https://www.genomicsengland.co.uk/initiatives/newborns/choosing-conditions?chapter=principle-b (accessed on 25 October 2024).
- Kaufmann, W.E.; Percy, A.K.; Neul, J.L.; Downs, J.; Leonard, H.; Nues, P.; Sharma, G.D.; Bartolotta, T.E.; Townend, G.S.; Curfs, L.M.G.; et al. Burden of illness in Rett syndrome: Initial evaluation of a disorder-specific caregiver survey. Orphanet J. Rare Dis. 2024, 19, 296. [Google Scholar] [CrossRef]
- Cianfaglione, R.; Hastings, R.P.; Felce, D.; Clarke, A.; Kerr, M.P. Psychological Well-Being of Mothers and Siblings in Families of Girls and Women with Rett Syndrome. J. Autism Dev. Disord. 2015, 45, 2939–2946. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Downs, J.; Wong, K.; Leonard, H. Longitudinal effects of caregiving on parental well-being: The example of Rett syndrome, a severe neurological disorder. Eur. Child Adolesc. Psychiatry 2019, 28, 505–520. [Google Scholar] [CrossRef]
- Killian, J.T., Jr.; Lane, J.B.; Lee, H.-S.; Pelham, J.H.; Skinner, S.A.; Kaufmann, W.E.; Glaze, D.G.; Neul, J.L.; Percy, A.K. Caretaker Quality of Life in Rett Syndrome: Disorder Features and Psychological Predictors. Pediatr. Neurol. 2016, 58, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Corchón, S.; Carrillo-López, I.; Cauli, O. Quality of life related to clinical features in patients with Rett syndrome and their parents: A systematic review. Metab. Brain Dis. 2018, 33, 1801–1810. [Google Scholar] [CrossRef]
- Marschik, P.B.; Lemcke, S.; Einspieler, C.; Zhang, D.; Bölte, S.; Townend, G.S.; Lauritsen, M.B. Early development in Rett syndrome—The benefits and difficulties of a birth cohort approach. Dev. Neurorehabilit. 2018, 21, 68–72. [Google Scholar] [CrossRef]
- Achilly, N.P.; Wang, W.; Zoghbi, H.Y. Presymptomatic training mitigates functional deficits in a mouse model of Rett syndrome. Nature 2021, 592, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Sutton, V.R.; Van den Veyver, I.B. Newborn screening and prenatal diagnosis for Rett syndrome: Implications for therapy. J. Child Neurol. 2005, 20, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.; Rodger, J.; Li, C.; Tan, X.; Hu, N.; Wong, K.; de Klerk, N.; Leonard, H. Environmental enrichment intervention for Rett syndrome: An individually randomised stepped wedge trial. Orphanet J. Rare Dis. 2018, 13, 3. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Mei, L.; Li, H.; Hu, C.; Zhou, B.; Zhang, K.; Qiao, Z.; Xu, X.; Xu, Q. Brain structural alterations in young girls with Rett syndrome: A voxel-based morphometry and tract-based spatial statistics study. Front. Neuroinform. 2022, 16, 962197. [Google Scholar] [CrossRef]
- Dragoumi, P.; O’Callaghan, F.; Zafeiriou, D.I. Diagnosis of tuberous sclerosis complex in the fetus. Eur. J. Paediatr. Neurol. 2018, 22, 1027–1034. [Google Scholar] [CrossRef]
- Sciacca, P.; Giacchi, V.; Mattia, C.; Greco, F.; Smilari, P.; Betta, P.; Distefano, G. Rhabdomyomas and Tuberous sclerosis complex: Our experience in 33 cases. BMC Cardiovasc. Disord. 2014, 14, 66. [Google Scholar] [CrossRef]
- Curatolo, P.; Specchio, N.; Aronica, E. Advances in the genetics and neuropathology of tuberous sclerosis complex: Edging closer to targeted therapy. Lancet Neurol. 2022, 21, 843–856. [Google Scholar] [CrossRef]
- Cosentino, L.; Vigli, D.; Franchi, F.; Laviola, G.; De Filippis, B. Rett syndrome before regression: A time window of overlooked opportunities for diagnosis and intervention. Neurosci. Biobehav. Rev. 2019, 107, 115–135. [Google Scholar] [CrossRef] [PubMed]
- Einspieler, C.; Freilinger, M.; Marschik, P.B. Behavioural biomarkers of typical Rett syndrome: Moving towards early identification. Wien. Med. Wochenschr. 2016, 166, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Einspieler, C.; Kerr, A.M.; Prechtl, H.F. Abnormal general movements in girls with Rett disorder: The first four months of life. Brain Dev. 2005, 27 (Suppl. S1), S8–S13. [Google Scholar] [CrossRef] [PubMed]
- Einspieler, C.; Kerr, A.M.; Prechtl, H.F. Is the early development of girls with Rett disorder really normal? Pediatr. Res. 2005, 57 Pt 1, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Roche, L.; Zhang, D.; Bartl-Pokorny, K.D.; Pokorny, F.B.; Schuller, B.W.; Esposito, G.; Bölte, S.; Roeyers, H.; Poustka, L.; Gugatschka, M.; et al. Early Vocal Development in Autism Spectrum Disorder, Rett Syndrome, and Fragile X Syndrome: Insights from Studies Using Retrospective Video Analysis. Adv. Neurodev. Disord. 2018, 2, 49–61. [Google Scholar] [CrossRef]
- Zhang, D.; Roche, L.; Bartl-Pokorny, K.D.; Krieber, M.; McLay, L.; Bolte, S.; Poustka, L.; Sigafoos, J.; Gugatschka, M.; Einspieler, C.; et al. Response to name and its value for the early detection of developmental disorders: Insights from autism spectrum disorder, Rett syndrome, and fragile X syndrome. A perspectives paper. Res. Dev. Disabil. 2018, 82, 95–108. [Google Scholar] [CrossRef]
- Percy, A.K.; Ananth, A.; Neul, J.L. Rett Syndrome: The Emerging Landscape of Treatment Strategies. CNS Drugs 2024, 38, 851–867. [Google Scholar] [CrossRef]
- Parent, H.; Ferranti, A.; Niswender, C. Trofinetide: A pioneering treatment for Rett syndrome. Trends Pharmacol. Sci. 2023, 44, 740–741. [Google Scholar] [CrossRef]
- Singh, J.; Santosh, P. Key issues in Rett syndrome: Emotional, behavioural and autonomic dysregulation (EBAD)—A target for clinical trials. Orphanet J. Rare Dis. 2018, 13, 128. [Google Scholar] [CrossRef]
- Hudu, S.A.; Elmigdadi, F.; Qtaitat, A.A.; Almehmadi, M.; Alsaiari, A.A.; Allahyani, M.; Aljuaid, A.; Salih, M.; Alghamdi, A.; Alrofaidi, M.A.; et al. Trofinetide for Rett Syndrome: Highlights on the Development and Related Inventions of the First USFDA-Approved Treatment for Rare Pediatric Unmet Medical Need. J. Clin. Med. 2023, 12, 5114. [Google Scholar] [CrossRef]
- A Novel, Regulated Gene Therapy (NGN-401) Study for Female Children with Rett Syndrome. Available online: https://clinicaltrials.gov/study/NCT05898620?cond=Rett%20Syndrome&term=neurogene&rank=1 (accessed on 1 November 2024).
- Safety and Efficacy of TSHA-102 in Adolescent and Adult Females with Rett Syndrome (REVEAL Adult Study). Available online: https://clinicaltrials.gov/study/NCT05606614?cond=Rett%20Syndrome&term=taysha&rank=1 (accessed on 1 November 2024).
- Safety and Efficacy of TSHA-102 in Pediatric Females with Rett Syndrome (REVEAL Pediatric Study). Available online: https://clinicaltrials.gov/study/NCT06152237?cond=Rett%20Syndrome&term=taysha&rank=2 (accessed on 1 November 2024).
- Singh, J.; Goodman-Vincent, E.; Santosh, P. Evidence Synthesis of Gene Therapy and Gene Editing from Different Disorders-Implications for Individuals with Rett Syndrome: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 9023. [Google Scholar] [CrossRef] [PubMed]
- The Association of the British Pharmaceutical Industry (ABPI), 2024: Unlocking Access to Future ATMPs in the UK Comparing International Approaches. Available online: https://www.abpi.org.uk/publications/unlocking-access-to-future-atmps-in-the-uk/ (accessed on 26 October 2024).
- Mahase, E. NICE rejects Alzheimer’s drug donanemab owing to cost and “significant health risks”. BMJ 2024, 387, q2342. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.N.; Bledsoe, J.R.; Grzywacz, B.; Beckman, A.; Bonner, M.; Eichler, F.S.; Kühl, J.S.; Harris, M.H.; Slauson, S.; Colvin, R.A.; et al. Hematologic Cancer after Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J Med. 2024, 391, 1287–1301. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.S. Digital Phenotyping in Clinical Neurology. Semin. Neurol. 2022, 42, 48–59. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Scahill, R.I.; Owen, G.; Durr, A.; Leavitt, B.R.; Roos, R.A.; Borowsky, B.; Landwehrmeyer, B.; Frost, C.; Johnson, H.; et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: Analysis of 36-month observational data. Lancet Neurol. 2013, 12, 637–649. [Google Scholar] [CrossRef]
- Blekher, T.; Johnson, S.A.; Marshall, J.; White, K.; Hui, S.; Weaver, M.; Gray, J.; Yee, R.; Stout, J.C.; Beristain, X.; et al. Saccades in presymptomatic and early stages of Huntington disease. Neurology 2006, 67, 394–399. [Google Scholar] [CrossRef]
- Ilg, W.; Fleszar, Z.; Schatton, C.; Hengel, H.; Harmuth, F.; Bauer, P.; Timmann, D.; Giese, M.; Schöls, L.; Synofzik, M. Individual changes in preclinical spinocerebellar ataxia identified via increased motor complexity. Mov. Disord. 2016, 31, 1891–1900. [Google Scholar] [CrossRef]
- Rusz, J.; Krack, P.; Tripoliti, E. From prodromal stages to clinical trials: The promise of digital speech biomarkers in Parkinson’s disease. Neurosci. Biobehav. Rev. 2024, 167, 105922. [Google Scholar] [CrossRef]
- Reece, A.G.; Danforth, C.M. Instagram photos reveal predictive markers of depression. EPJ Data Sci. 2017, 6, 15. [Google Scholar] [CrossRef]
- Bedi, G.; Carrillo, F.; Cecchi, G.A.; Slezak, D.F.; Sigman, M.; Mota, N.B.; Ribeiro, S.; Javitt, D.C.; Copelli, M.; Corcoran, C.M. Automated analysis of free speech predicts psychosis onset in high-risk youths. npj Schizophr. 2015, 1, 15030. [Google Scholar] [CrossRef]
- Perochon, S.; Di Martino, J.M.; Carpenter, K.L.H.; Compton, S.; Davis, N.; Eichner, B.; Espinosa, S.; Franz, L.; Babu, P.R.K.; Sapiro, G.; et al. Early detection of autism using digital behavioral phenotyping. Nat. Med. 2023, 29, 2489–2497. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Lanzarini, E.; Santosh, P. Autonomic Characteristics of Sudden Unexpected Death in Epilepsy in Children—A Systematic Review of Studies and Their Relevance to the Management of Epilepsy in Rett Syndrome. Front. Neurol. 2021, 11, 632510. [Google Scholar] [CrossRef] [PubMed]
- Petriti, U.; Dudman, D.C.; Scosyrev, E.; Lopez-Leon, S. Global prevalence of Rett syndrome: Systematic review and meta-analysis. Syst. Rev. 2023, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Mishra, G.P.; Sun, E.X.; Chin, T.; Eckhardt, M.; Greenberg, M.E.; Stroud, H. Interaction of methyl-CpG-binding protein 2 (MeCP2) with distinct enhancers in the mouse cortex. Nat. Neurosci. 2024. [Google Scholar] [CrossRef]
- Brower, A.; Chan, K.; Williams, M.; Berry, S.; Currier, R.; Rinaldo, P.; Caggana, M.; Gaviglio, A.; Wilcox, W.; Steiner, R.; et al. Population-Based Screening of Newborns: Findings from the NBS Expansion Study (Part One). Front. Genet. 2022, 13, 867337. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, J.; Santosh, P. The Newborn Screening Programme Revisited: An Expert Opinion on the Challenges of Rett Syndrome. Genes 2024, 15, 1570. https://doi.org/10.3390/genes15121570
Singh J, Santosh P. The Newborn Screening Programme Revisited: An Expert Opinion on the Challenges of Rett Syndrome. Genes. 2024; 15(12):1570. https://doi.org/10.3390/genes15121570
Chicago/Turabian StyleSingh, Jatinder, and Paramala Santosh. 2024. "The Newborn Screening Programme Revisited: An Expert Opinion on the Challenges of Rett Syndrome" Genes 15, no. 12: 1570. https://doi.org/10.3390/genes15121570
APA StyleSingh, J., & Santosh, P. (2024). The Newborn Screening Programme Revisited: An Expert Opinion on the Challenges of Rett Syndrome. Genes, 15(12), 1570. https://doi.org/10.3390/genes15121570